核壳结构光催化剂及其制备方法和应用与流程
本发明属于可见光光催化
技术领域:
,具体涉及一种核壳结构光催化剂及其制备方法和应用。
背景技术:
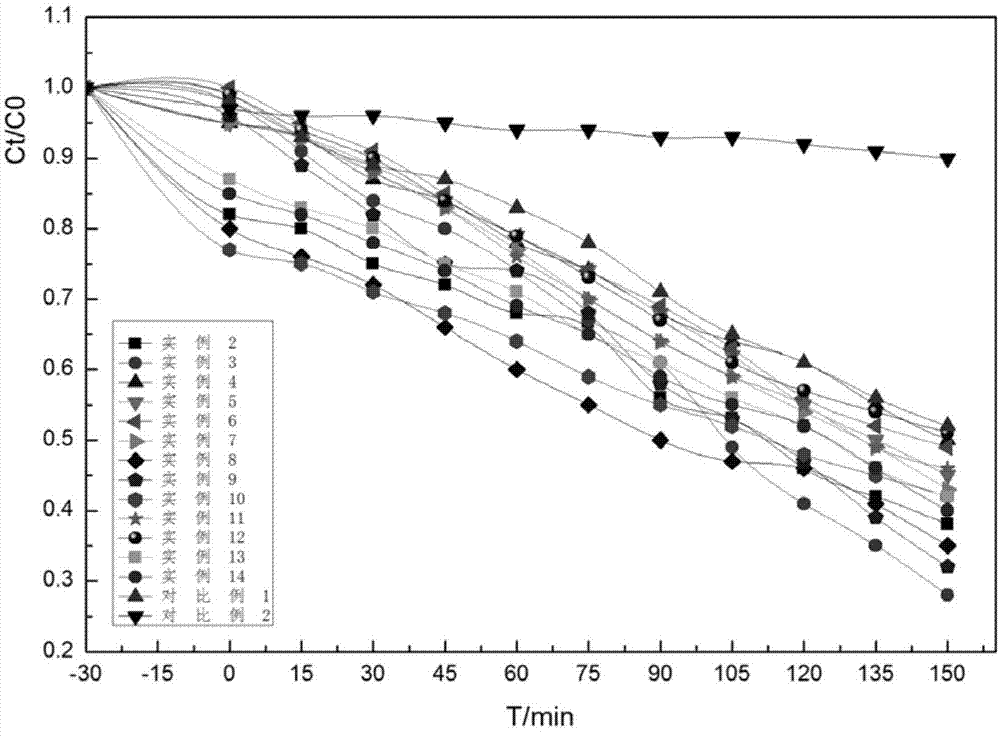
随着全球环境污染日益严重,环境问题越来越受到人们的关注。半导体光催化剂作为一种新的催化材料,在污染物处理领域有着广阔的应用前景。目前,国内外应用最广泛的半导体光催化剂是tio2,但是它属于宽禁带半导体,仅仅只能在紫外线光(仅占太阳辐射的4%)照射下才能产生光催化活性,这在很大程度上限制了tio2的应用,且粉末状tio2在使用过程中存在分离回收困难的问题。近年来光催化剂研究开始通过开发具有可见光响应的新型光催化材料,然而这种催化剂往往存在电子-空穴分离率低、复合率高,且难以回收再利用,增加了使用成本。而且,这种光催化剂难以回收再利用,增加了使用成本。因此,开发具有可见光响应、降低电子-空穴复合率、易于回收的光催化剂变成急需解决的问题。鉴于此,特提出本发明。技术实现要素:本发明的目的在于提供一种核壳结构光催化剂;该光催化剂具有可见光响应,较优的电子-空穴复合率,可循环使用。本发明的另一目的在于提供上述核壳结构光催化剂的制备方法;整个工艺简单,适用于大规模生产,且经济环保。本发明的目的还在于提供上述核壳结构光催化剂在降解有机污染物中的应用;该光催化剂在可见光下光降解有机污染物具有很高的稳定性和降解率,循环5次以上而无明显活性下降,可循环利用。根据本发明的一个方面,本发明提供一种核壳结构光催化剂,所述光催化剂以fe3o4为核,fe3o4核外包覆nial-ldhs层形成fe3o4@ldhs,fe3o4@ldhs外包覆有bi2moo6层。作为本发明的一种优选实施方式,所述fe3o4与nial-ldhs的重量比为0.35-1:2.5-5;所述fe3o4@ldhs与bi2moo6的重量比为1-2:2.5-5;优选地,所述fe3o4与nial-ldhs的重量比为1:2.5-5;所述fe3o4@ldhs与bi2moo6的重量比为1-1.5:2.5。根据本发明的另一个方面,本发明提供所述的核壳结构光催化剂的制备方法,包括以下步骤:(a)向溶剂中加入fe3o4、含ni2+和al3+的离子溶液和碱溶液,在150-170℃下进行水热反应,得到fe3o4@ldhs;(b)向fe3o4@ldhs中加入bi(no3)3和钼酸或钼酸盐,在ph为9-10的条件下,使其发生水热反应,得到核壳结构光催化剂。作为本发明的一种优选实施方式,将fecl3·h2o、弱酸性氧化剂和表面活性剂加入溶剂中,使其发生溶剂热反应,得到fe3o4。作为本发明的一种优选实施方式,所述fecl3·h2o、弱酸性氧化剂、表面活性剂和溶剂的投料比为1-1.5:3.5-4:0.8-1.2:50-70g/g/g/ml,优选为1.35:3.6:1:60g/g/g/ml;优选地,所述弱酸性氧化剂为醋酸盐,优选为naac或kac;优选地,所述表面活性剂为非离子型活性剂或阴离子表面活性剂;优选地,所述非离子型活性剂为聚乙二醇;优选地,所述阴离子表面活性剂为十二烷基三甲基溴化铵或十二烷基苯磺酸钠;优选地,所述表面活性剂为聚乙二醇;优选地,所述溶剂为c2-c6的多元醇;优选地,所述c2-c6的多元醇为乙二醇、二甘醇、三甘醇或甘露醇,优选为乙二醇;优选地,所述溶剂热反应的温度为180-220℃,优选为200℃;优选地,所述溶剂热反应的时间为8-12h,优选为10h;优选地,所述方法还包括待溶剂热反应完全后依次进行分离和干燥得到fe3o4的步骤。作为本发明的一种优选实施方式,步骤(a)中,所述含ni2+和al3+的离子溶液中,ni2+离子浓度为0.3-0.5mol/l,优选为0.4mol/l,al3+离子浓度为0.15-0.25mol/l,优选为0.2mol/l;优选地,所述含ni2+和al3+的离子溶液均独立地为ni2+和al3+的可溶性盐形成的水溶液;优选地,所述ni2+和al3+的可溶性盐均独立地为硝酸盐、硫酸盐、盐酸盐或溴酸盐;优选地,所述碱溶液为六亚甲基四胺水溶液、尿素水溶液或氨水;优选为六亚甲基四胺水溶液,进一步优选为浓度为0.8-1.2mol/l的六亚甲基四胺水溶液,最优选为浓度为1mol/l的六亚甲基四胺水溶液;优选地,所述fe3o4、ni2+离子和al3+离子的投料比为35-150:2-10:1-5g/mol/mol,优选为80-120:4-8:2-4g/mol/mol;优选地,所述fe3o4与碱溶液中碱的重量摩尔比为35-150:5-25g/mol,优选为80-120:10-20g/mol;优选地,步骤(a)中,所述水热反应的温度为160℃;优选地,步骤(a)中,所述水热反应的时间为45-50h;优选地,步骤(a)中,所述方法还包括待水热反应完全后依次进行分离和干燥得到fe3o4@ldhs的步骤。作为本发明的一种优选实施方式,步骤(b)中,所述bi(no3)3以bi(no3)3水溶液的形式加入,优选以浓度为0.35-0.45mol/l的bi(no3)3水溶液的形式加入,进一步优选以浓度为0.4mol/l的bi(no3)3水溶液的形式加入;优选地,所述钼酸或钼酸盐以钼酸或钼酸盐水溶液的形式加入,优选以浓度为0.18-0.22mol/l的钼酸或钼酸盐水溶液的形式加入,进一步优选以浓度为0.2mol/l的钼酸或钼酸盐水溶液的形式加入;优选地,所述钼酸盐为钼酸钠或钼酸钾;优选地,所述fe3o4@ldhs、bi(no3)3与钼酸或钼酸盐的用量比为100-200:2-10:1-5g/mol/mol,优选为120-180:4-8:2-4g/mol/mol;作为本发明的一种优选实施方式,步骤(b)中,所述水热反应的温度为150-170℃,优选为160℃;步骤(b)中,所述水热反应的时间为45-50h,优选为48h;优选地,步骤(b)中,所述方法还包括待水热反应完全后依次进行分离和干燥得到核壳结构光催化剂的步骤。作为本发明的一种优选实施方式,所述的制备方法包括以下步骤:(a)向溶剂中加入fe3o4、含ni2+和al3+的离子溶液和碱溶液,在150-170℃下进行水热反应,得到fe3o4@ldhs;(b)向fe3o4@ldhs中加入bi(no3)3和钼酸或钼酸盐,在ph为9-10的条件下,使其发生水热反应,得到核壳结构光催化剂;步骤(a)中,所述含ni2+和al3+的离子溶液中,ni2+离子浓度为0.3-0.5mol/l,优选为0.4mol/l,al3+离子浓度为0.15-0.25mol/l,优选为0.2mol/l;优选地,所述含ni2+和al3+的离子溶液均独立地为ni2+和al3+的可溶性盐形成的水溶液;优选地,所述ni2+和al3+的可溶性盐均独立地为硝酸盐、硫酸盐、盐酸盐或溴酸盐;优选地,所述碱溶液为六亚甲基四胺水溶液、尿素水溶液或氨水;优选为六亚甲基四胺水溶液,进一步优选为浓度为0.8-1.2mol/l的六亚甲基四胺水溶液,最优选为浓度为1mol/l的六亚甲基四胺水溶液;优选地,所述fe3o4、ni2+离子和al3+离子的投料比为35-150:2-10:1-5g/mol/mol,优选为80-120:4-8:2-4g/mol/mol;优选地,所述fe3o4与碱溶液中碱的重量摩尔比为35-150:5-25g/mol,优选为80-120:10-20g/mol;优选地,步骤(a)中,所述水热反应的温度为160℃;优选地,步骤(a)中,所述水热反应的时间为45-50h;优选地,步骤(a)中,所述方法还包括待水热反应完全后依次进行分离和干燥得到fe3o4@ldhs的步骤;步骤(b)中,所述bi(no3)3以bi(no3)3水溶液的形式加入,优选以浓度为0.35-0.45mol/l的bi(no3)3水溶液的形式加入,进一步优选以浓度为0.4mol/l的bi(no3)3水溶液的形式加入;优选地,所述钼酸或钼酸盐以钼酸或钼酸盐水溶液的形式加入,优选以浓度为0.18-0.22mol/l的钼酸或钼酸盐水溶液的形式加入,进一步优选以浓度为0.2mol/l的钼酸或钼酸盐水溶液的形式加入;优选地,所述钼酸盐为钼酸钠或钼酸钾;优选地,所述fe3o4@ldhs、bi(no3)3与钼酸或钼酸盐的用量比为100-200:2-10:1-5g/mol/mol,优选为120-180:4-8:2-4g/mol/mol;优选地,步骤(b)中,所述水热反应的温度为150-170℃,优选为160℃;步骤(b)中,所述水热反应的时间为45-50h,优选为48h;优选地,步骤(b)中,所述方法还包括待水热反应完全后依次进行分离和干燥得到核壳结构光催化剂的步骤。根据本发明的另一个方面,本发明提供所述的核壳结构光催化剂在光降解有机污染物中的应用。作为本发明的一种优选实施方式,所述光降解有机污染物的光是可见光或紫外光;优选地,所述有机污染物为有机染料;优选地,所述有机染料为亚甲基蓝、罗丹明或甲基橙。本发明提供了一种核壳结构光催化剂,所述光催化剂以fe3o4为核,改善水滑石堆积、叠加的现状,使层板分散,增加比表面积,暴露更多的活性位点;另外,以磁性fe3o4为核芯,可以解决光催化剂的回收问题,使得本发明核壳结构光催化剂可循环利用,循环5次以上而活性无明显下降,降低成本。该光催化剂的fe3o4核外包覆nial-ldhs层形成fe3o4@ldhs,fe3o4@ldhs外包覆有bi2moo6层,使得nial-ldhs与bi2moo6复合形成异质结材料,利用水滑石的高分散性和高吸附性,改善bi2moo6的光催化性能,增强其光催化活性;另外,nial-ldhs与bi2moo6复合可以调节二者的禁带宽度,使其具有紫外-可见双重响应,更有效地改善光生空穴与电子的复合率。本发明光催化剂在可见光下光降解有机污染物具有很高的稳定性和降解率。附图说明为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。图1为本发明实施例1制备的fe3o4@ldhs的ftir谱图;图2为本发明实施例1制备的核壳结构光催化剂fe3o4@ldhs-bi2moo6的ftir谱图;图3为本发明实施例1制备的fe3o4@ldhs的xrd谱图;图4为本发明实施例1制备的核壳结构光催化剂fe3o4@ldhs-bi2moo6的xrd谱图;图5为本发明实施例1制备的核壳结构光催化剂fe3o4@ldhs-bi2moo6的xrd放大后谱图;图6为本发明实施例1制备的核壳结构光催化剂fe3o4@ldhs-bi2moo6的漫反射谱图(a:bi2moo6、b:ldhs-bi2moo6、c:ldhs);图7为本发明实施例1制备的fe3o4@ldhs的sem谱图;图8为本发明实施例1制备的核壳结构光催化剂fe3o4@ldhs-bi2moo6的sem图;图9为本发明试验例1光催化测试的结果;图10为本发明试验例3催化剂降解的循环实验。具体实施方式下面将结合实施例及附图对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。根据本发明的一个方面,本发明提供一种核壳结构光催化剂,所述光催化剂以fe3o4为核,fe3o4核外包覆nial-ldhs层形成fe3o4@ldhs,fe3o4@ldhs外包覆有bi2moo6层。本发明中,“fe3o4@ldhs”为fe3o4核外包覆nial-ldhs层形成的核壳结构的缩写。本发明中,“fe3o4@ldhs外包覆有bi2moo6层”形成核壳结构光催化剂,核壳结构光催化剂的缩写为“fe3o4@ldhs-bi2moo6”。本发明核壳结构光催化剂以fe3o4为核,一方面可以改善水滑石堆积、叠加的现状,使层板分散,增加了比表面积,将会暴露更多的活性位点;另一方面核壳结构光催化剂以磁性fe3o4为核芯,可以解决光催化剂的回收问题,使得本发明核壳结构光催化剂可循环利用,循环5次以上而活性无明显下降,降低成本。该核壳结构光催化剂的fe3o4核外包覆nial-ldhs层形成fe3o4@ldhs,fe3o4@ldhs外包覆有bi2moo6层,使得nial-ldhs与bi2moo6复合形成异质结材料,利用水滑石的高分散性和高吸附性,改善bi2moo6的光催化性能,增强其光催化活性;另外,nial-ldhs与bi2moo6复合可以调节二者的禁带宽度,使其具有紫外-可见双重响应,更有效地改善光生空穴与电子的复合率。本发明核壳结构光催化剂在可见光下光降解有机污染物具有很高的稳定性和降解率。作为本发明的一种优选实施方式,fe3o4与nial-ldhs的重量比为0.35-1:2.5-5,优选为1:2.5-5。其中,按重量计,fe3o4典型但非限制性的重量份为0.35份、0.4份、0.45份、0.5份、0.6份、0.7份、0.75份、0.8份、0.85份、0.9份、0.95份或1份等;nial-ldhs典型但非限制性的重量份为2.5份、2.6份、2.8份、3份、3.2份、3.5份、3.8份、4份、4.2份、4.5份或5份等。作为本发明的一种优选实施方式,fe3o4@ldhs与bi2moo6的重量比为1-2:2.5-5,优选为1-1.5:2.5。其中,按重量计,fe3o4@ldhs典型但非限制性的重量份为1份、1.1份、1.2份、1.25份、1.3份、1.4份、1.5份、1.6份、1.7份、1.8份或2份等;bi2moo6典型但非限制性的重量份为2.5份、2.6份、2.8份、3份、3.2份、3.5份、3.8份、4份、4.2份、4.5份或5份等。作为本发明的一种优选实施方式,所述fe3o4与nial-ldhs的重量比为0.35-1:2.5-5;所述fe3o4@ldhs与bi2moo6的重量比为1-2:2.5-5。在本发明的优选实施方式中,本发明实施例2-11、实施例13和14通过调整各原料的用量,使得fe3o4与nial-ldhs的重量比为0.35-1:2.5-5,fe3o4@ldhs与bi2moo6的重量比为1-2:2.5-5,进一增强其光催化活性,在光催化实验中,经过150min后,实施例2-11、实施例13和14的亚甲基蓝剩余量都在50%以下,得到的光催化剂在兼具成本的同时具有很高的稳定性和降解率。作为本发明的一种优选实施方式,所述fe3o4与nial-ldhs的重量比为1:2.5-5;所述fe3o4@ldhs与bi2moo6的重量比为1-1.5:2.5。在本发明的优选实施方式中,本发明实施例2、3、8、10通过调整fe3o4与nial-ldhs的重量比为1:2.5-5,以及调整fe3o4@ldhs与bi2moo6的重量比为1-1.5:2.5,进一步增强其光催化活性,在光催化实验中,经过150min后,实施例2、3、8、10的亚甲基蓝剩余量都在42%以下,尤其是实施例3中亚甲基蓝只剩余28%,得到的光催化剂在兼具成本的同时具有很高的稳定性和降解率。根据本发明的另一个方面,本发明提供所述的核壳结构光催化剂的制备方法,包括以下步骤:(a)向溶剂中加入fe3o4、含ni2+和al3+的离子溶液和碱溶液,在150-170℃下进行水热反应,得到fe3o4@ldhs;(b)向fe3o4@ldhs中加入bi(no3)3和钼酸或钼酸盐,在ph为9-10的条件下,使其发生水热反应,得到核壳结构光催化剂。本发明通过两步水热反应制备得到核壳结构光催化剂,工艺简单,适用于大规模生产,且制备过程经济环保。需要说明的是,本发明对于fe3o4、含ni2+和al3+的离子溶液、碱溶液、bi(no3)3和钼酸或钼酸盐的来源没有特殊的限制,采用本领域技术人员所熟知的各原料即可;如可以采用其市售商品,也可以采用本领域技术人员熟知的制备方法自行制备。本发明中,“含ni2+和al3+的离子溶液”可以是指在一种溶液中同时含有ni2+和al3+两种离子;也可以是指两种分别含有ni2+和al3+的离子溶液。步骤(a)中,所述fe3o4可以是通用的可作为光催化剂核的fe3o4,在本发明的一些优选实施方式中,所述fe3o4也可以是由包括以下步骤的方法制备得到的:将fecl3·h2o、弱酸性氧化剂和表面活性剂加入溶剂中,使其发生溶剂热反应,得到fe3o4。本发明中乙二醇为有机溶剂,而我们制备的物质属于无机物,表面活性剂的存在可使得两者更好的相容,改变了物质的表面张力,增大接触角,使二者之间接触更充分。作为本发明的一种优选实施方式,所述fecl3·h2o、弱酸性氧化剂、表面活性剂和溶剂的投料比为1-1.5:3.5-4:0.8-1.2:50-70g/g/g/ml,优选为1.35:3.6:1:60g/g/g/ml。按重量体积份计,fecl3·h2o的重量份为1-1.5,优选为1.35,典型但非限制性的重量份为1份、1.1份、1.2份、1.25份、1.3份、1.35份、1.4份或1.5份等;弱酸性氧化剂的重量份为3.5-4,优选为3.6,典型但非限制性的重量份为3.5份、3.6份、3.7份、3.8份或4份等;表面活性剂的重量份为0.8-1.2,优选为1,典型但非限制性的重量份为0.8份、0.9份、1份、1.1份或1.2份;溶剂的体积份为50-70,优选为60,典型但非限制性的体积份为50份、51份、53份、55份、56份、56份、58份59、60份、61份、63份、65份、66份、68份或70份等;上述重量体积比单位是g/ml。作为本发明的一种优选实施方式,所述弱酸性氧化剂为醋酸盐,优选为naac或kac;本发明弱酸性氧化剂可避免制备得到的是fe3o4和fe2o3的混合物,使得制备得到的为纯的fe3o4,具体的,可以选择naac或kac作为弱酸性氧化剂。作为本发明的一种优选实施方式,所述表面活性剂为非离子型活性剂或阴离子表面活性剂;所述非离子型活性剂可以为聚乙二醇;所述阴离子表面活性剂可以为十二烷基三甲基溴化铵或十二烷基苯磺酸钠。作为本发明的一种优选实施方式,所述表面活性剂为聚乙二醇。作为本发明的一种优选实施方式,所述溶剂为c2-c6的多元醇。优选地,所述c2-c6的多元醇为乙二醇、二甘醇、三甘醇或甘露醇;优选为乙二醇。在本发明的优选实施方式中,表面活性剂聚乙二醇是溶剂乙二醇的聚合物,解决不同溶剂之间分层的问题,使溶剂形成一种均一稳定的状态。另外,聚合物的加入使四氧化三铁更好的分散,以及利用表面张力的作用生成更均一的颗粒。作为本发明的一种优选实施方式,所述溶剂热反应的温度为180-220℃,优选为200℃;所述溶剂热反应的时间为8-12h,优选为10h。在本发明的优选实施方式中,通过调整溶剂热反应合适的温度和时间,成功制备得到了fe3o4。作为本发明的一种优选实施方式,所述方法还包括待溶剂热反应完全后依次进行分离和干燥得到fe3o4的步骤。在本发明的优选实施方式中,所述分离包括将反应完全后得到的液体依次进行离心和洗涤;所述离心的转速为80000-120000r/min,所述离心的时间为2-4min;所述洗涤是依次用水洗涤三次和用乙醇洗涤一次。在本发明的优选实施方式中,通过调整合适的反应时间、温度等反应条件,制备得到fe3o4。作为本发明的一种优选实施方式,步骤(a)中,所述含ni2+和al3+的离子溶液中,ni2+离子浓度为0.3-0.5mol/l,优选为0.4mol/l,al3+离子浓度为0.15-0.25mol/l,优选为0.2mol/l。所述含ni2+和al3+的离子溶液均独立地为ni2+和al3+的可溶性盐形成的水溶液;含ni2+和al3+的离子溶液可以是有ni2+和al3+的任何可溶性盐配置成的溶液;优选地,所述ni2+和al3+的可溶性盐均独立地为硝酸盐、硫酸盐、盐酸盐或溴酸盐,其中,ni2+的可溶性盐为硝酸镍、硫酸镍、盐酸镍或溴酸镍,al3+的可溶性盐为硝酸铝、硫酸铝、盐酸铝或溴酸铝。优选地,所述碱溶液为六亚甲基四胺水溶液、尿素水溶液或氨水;优选为六亚甲基四胺水溶液,进一步优选为浓度为0.8-1.2mol/l的六亚甲基四胺水溶液,最优选为浓度为1mol/l的六亚甲基四胺水溶液。为了控制水滑石的成核速率和生成速率,使生长速率大于成核速率得到更大粒径的层板结构,我们需要一种缓慢变成碱性的化学环境。在本发明的优选实施方式中,六亚甲基四胺会在高温下分解成氨气和二氧化碳,进而使溶液缓慢变成碱性。同时二氧化碳溶解水中形成碳酸根离子进而成为水滑石的层间离子。优选地,所述fe3o4、ni2+离子和al3+离子的投料比为35-150:2-10:1-5g/mol/mol,优选为80-120:4-8:2-4g/mol/mol;按重量摩尔份计,重量摩尔比为g/mol,fe3o4典型但非限制性的重量份为35份、40份、45份、50份、55份、60份、65份、70份、75份、80份、85份、90份、95份、100份、105份、110份、120份、130份、140份或150份等;ni2+离子典型但非限制性的摩尔份为2份、3份、4份、5份、6份、7份、8份、9份或10份等;al3+离子典型但非限制性的摩尔份为1份、2份、3份、4份或5份等。此处,ni2+离子和al3+离子的量通过离子的浓度来限定计量比。优选地,所述fe3o4与碱溶液中碱的重量摩尔比为35-150:5-25g/mol,优选为80-120:10-20g/mol;按重量摩尔份计,重量摩尔比为g/mol,fe3o4典型但非限制性的重量份为35份、40份、45份、50份、55份、60份、65份、70份、75份、80份、85份、90份、95份、100份、105份、110份、120份、130份、140份或150份等;碱典型但非限制性的摩尔份为5份、6份、8份、9份、10份、12份、15份、18份、20份、22份或25份等。通过合理调整fe3o4、ni2+离子、al3+离子和碱原料用量之间的配比,制备得到fe3o4@ldhs。优选地,步骤(a)中,所述水热反应的温度为160℃;所述水热反应的时间为45-50h。在本发明的优选实施方式中,通过选择合适的反应温度和时间等反应条件,成功制备得到了fe3o4核外包覆nial-ldhs层形成的fe3o4@ldhs。优选地,步骤(a)中,所述方法还包括待水热反应完全后依次进行分离和干燥得到fe3o4@ldhs的步骤。在本发明的优选实施方式中,所述分离包括将反应完全后得到的液体依次进行离心和洗涤;所述离心的转速为80000-120000r/min,所述离心的时间为2-4min;所述洗涤是依次用水洗涤三次和用乙醇洗涤一次。作为本发明的一种优选实施方式,步骤(b)中,所述bi(no3)3以bi(no3)3水溶液的形式加入,优选以浓度为0.35-0.45mol/l的bi(no3)3水溶液的形式加入,进一步优选以浓度为0.4mol/l的bi(no3)3水溶液的形式加入;bi(no3)3水溶液典型但非限制性的浓度为0.35mol/l、0.36mol/l、0.37mol/l、0.38mol/l、0.39mol/l、0.40mol/l、0.41mol/l、0.42mol/l、0.43mol/l、0.44mol/l或0.45mol/l。作为本发明的一种优选实施方式,所述钼酸或钼酸盐以钼酸或钼酸盐水溶液的形式加入,优选以浓度为0.18-0.22mol/l的钼酸或钼酸盐水溶液的形式加入,进一步优选以浓度为0.2mol/l的钼酸或钼酸盐水溶液的形式加入;钼酸或钼酸盐水溶液典型但非限制性的浓度为0.18mol/l、0.19mol/l、0.20mol/l、0.41mol/l或0.22mol/l。优选地,所述钼酸盐为钼酸钠或钼酸钾。作为本发明的一种优选实施方式,所述fe3o4@ldhs、bi(no3)3与钼酸或钼酸盐的用量比为100-200:2-10:1-5g/mol/mol,优选为120-180:4-8:2-4g/mol/mol。按重量摩尔份计,重量摩尔比为g/mol,fe3o4@ldhs典型但非限制性的重量份为100份、120份、140份、150份、160份、170份、180份或200份等;bi(no3)3典型但非限制性的摩尔份为2份、3份、4份、5份、6份、7份、8份、9份或10份等;钼酸或钼酸盐典型但非限制性的摩尔份为1份、2份、3份、4份或5份等。“fe3o4@ldhs、bi(no3)3与钼酸或钼酸盐的用量比为100-200:2-10:1-5g/mol/mol”是指fe3o4@ldhs为100-200重量份、bi(no3)3为2-10摩尔份、钼酸或钼酸盐为1-5摩尔份。作为本发明的一种优选实施方式,步骤(b)中,所述水热反应的温度为150-170℃,优选为160℃;步骤(b)中,所述水热反应的时间为45-50h,优选为48h。在本发明的优选实施方式中,通过选择合适的反应温度和时间等反应条件,更有利于成功制备得到fe3o4@ldhs外包覆bi2moo6层形成的光催化剂fe3o4@ldhs-bi2moo6。优选地,步骤(b)中,所述方法还包括待水热反应完全后依次进行分离和干燥得到核壳结构光催化剂的步骤。作为本发明的一种优选实施方式,所述分离包括离心和洗涤,所述离心的转速为80000-120000r/min,所述离心的时间为2-4min;所述洗涤是依次用水洗涤三次和用乙醇洗涤一次。作为本发明的一种优选实施方式,所述的制备方法包括以下步骤:(a)向溶剂中加入fe3o4、含ni2+和al3+的离子溶液和碱溶液,在150-170℃下进行水热反应,得到fe3o4@ldhs;(b)向fe3o4@ldhs中加入bi(no3)3和钼酸或钼酸盐,在ph为9-10的条件下,使其发生水热反应,得到核壳结构光催化剂。步骤(a)中,所述含ni2+和al3+的离子溶液中,ni2+离子浓度为0.3-0.5mol/l,优选为0.4mol/l,al3+离子浓度为0.15-0.25mol/l,优选为0.2mol/l。优选地,所述含ni2+和al3+的离子溶液均独立地为ni2+和al3+的可溶性盐形成的水溶液。优选地,所述ni2+和al3+的可溶性盐均独立地为硝酸盐、硫酸盐、盐酸盐或溴酸盐,其中,ni2+的可溶性盐为硝酸镍、硫酸镍、盐酸镍或溴酸镍,al3+的可溶性盐为硝酸铝、硫酸铝、盐酸铝或溴酸铝。优选地,所述碱溶液为六亚甲基四胺水溶液、尿素水溶液或氨水;优选为六亚甲基四胺水溶液,进一步优选为浓度为0.8-1.2mol/l的六亚甲基四胺水溶液,最优选为浓度为1mol/l的六亚甲基四胺水溶液。优选地,所述fe3o4、ni2+离子和al3+离子的投料比为35-150:2-10:1-5g/mol/mol,优选为80-120:4-8:2-4g/mol/mol。优选地,所述fe3o4与碱溶液中碱的重量摩尔比为35-150:5-25g/mol,优选为80-120:10-20g/mol。优选地,步骤(a)中,所述水热反应的温度为160℃。优选地,步骤(a)中,所述水热反应的时间为45-50h。优选地,步骤(a)中,所述方法还包括待水热反应完全后依次进行分离和干燥得到fe3o4@ldhs的步骤。步骤(b)中,所述bi(no3)3以bi(no3)3水溶液的形式加入,优选以浓度为0.35-0.45mol/l的bi(no3)3水溶液的形式加入,进一步优选以浓度为0.4mol/l的bi(no3)3水溶液的形式加入。优选地,所述钼酸或钼酸盐以钼酸或钼酸盐水溶液的形式加入,优选以浓度为0.18-0.22mol/l的钼酸或钼酸盐水溶液的形式加入,进一步优选以浓度为0.2mol/l的钼酸或钼酸盐水溶液的形式加入。优选地,所述钼酸盐为钼酸钠或钼酸钾。优选地,所述fe3o4@ldhs、bi(no3)3与钼酸或钼酸盐的用量比为100-200:2-10:1-5g/mol/mol,优选为120-180:4-8:2-4g/mol/mol。优选地,步骤(b)中,所述水热反应的温度为150-170℃,优选为160℃。步骤(b)中,所述水热反应的时间为45-50h,优选为48h。优选地,步骤(b)中,所述方法还包括待水热反应完全后依次进行分离和干燥得到核壳结构光催化剂的步骤。在本发明的优选实施方式中,通过调整fe3o4@ldhs、bi(no3)3水溶液和钼酸或钼酸盐的用量,以及反应温度、时间等反应条件,制备得到了fe3o4@ldhs外包覆bi2moo6层形成的核壳结构光催化剂fe3o4@ldhs-bi2moo6。fe3o4@ldhs外包覆有bi2moo6层,使得nial-ldhs与bi2moo6复合形成异质结材料,利用水滑石的高分散性和高吸附性,改善bi2moo6的光催化性能,增强其光催化活性;另外,nial-ldhs与bi2moo6复合可以调节二者的禁带宽度,使其具有紫外-可见双重响应,更有效地改善光生空穴与电子的复合率。根据本发明的另一个方面,本发明提供所述的核壳结构光催化剂在降解有机污染物中的应用。本发明光催化剂在可见光下光降解有机污染物具有很高的稳定性和降解率,循环5次以上而活性无明显下降,可循环利用,降低成本。作为本发明的一种优选实施方式,所述光降解有机污染物的光是可见光或紫外光。优选地,所述有机污染物为有机染料。优选地,所述有机染料为亚甲基蓝、罗丹明或甲基橙。本发明光催化剂具有紫外-可见双重响应,在可见光或紫外光下都可光降解有机污染物。需要说明的是,本发明反应中所述的室温并无特殊的限制,可以为5-35℃,优选为20-30℃;所述的室温典型但非限制性的温度为5℃、6℃、7℃、8℃、9℃、10℃、11℃、12℃、13℃、15℃、17℃、19℃、20℃、22℃、23℃、25℃、27℃、29℃、30℃、33℃或35℃等。下面将结合实施例和对比例对本发明的技术方案进行进一步地说明。实施例11、核壳结构光催化剂的合成(1)合成fe3o4称取1.35g的fecl3·h2o、3.6gnaac、1g聚乙二醇、60ml乙二醇放入容量为250ml的三口烧瓶中,在室温下超声混合15min后,将母液移至水热釜,放入烘箱中在200℃下水热10h。10h后从烘箱中取出水热釜并进行冷却,冷却至室温后,用离心机(10000r/min,3min)进行离心,用蒸馏水反复洗涤3次,然后再用乙醇洗涤一次,除去上层清液,将样品进行干燥;干燥后研磨成粉待用。(2)合成fe3o4@ldhs称取0.1g的fe3o4,用移液管分别移取20ml配置好的ni2+、al3+离子溶液(ni2+离子浓度为0.4m的硝酸镍水溶液和al3+离子浓度为0.2m的硝酸铝水溶液)和20ml浓度为1m的naoh水溶液,放入容量为250ml的三口烧瓶中,加入50ml蒸馏水,在室温下超声混合15min后将母液移至水热釜,放入160℃的烘箱中水热48h。48h后从烘箱中取出水热釜并进行冷却,冷却至室温后,用离心机(10000r/min,3min)进行离心,用蒸馏水反复洗涤3次,然后再用乙醇洗涤一次,除去上层清液并进行干燥,干燥后研磨成粉待用,得到fe3o4@ldhs。(3)合成核壳结构光催化剂fe3o4@ldhs-bi2moo6称取0.1g的fe3o4@ldhs放入100ml烧杯中,用移液管分别移取20ml的0.4m的bi(no3)3水溶液和20ml的0.2m的na2moo4溶液水,放入100ml烧杯中,室温超声搅拌30min之后转入250ml的三口烧瓶中,用磁力搅拌器一直搅拌并用ph测试仪来检测ph值(保持溶液的ph值为10,若小于10,则滴加0.1m的naoh溶液;若大于10,则滴加0.1m的hno3溶液)。滴加完毕后将母液移至水热釜,放入烘箱中在160℃下水热48h。48h后从烘箱中取出水热釜并进行冷却,冷却至室温后,用离心机(10000r/min,3min)进行离心,用蒸馏水反复洗涤3次,然后再用乙醇洗涤一次,除去上层清液并进行干燥,干燥后研磨成粉待用。2、表征图1和图2分别为为本发明实施例1制备的fe3o4@ldhs和fe3o4@ldhs-bi2moo6的ftir谱图;我们利用ft-ir分析方法来确定所制得的ldhs样品的层板羟基、层间阴离子的排布情况及其种类,在图1中在3424cm-1左右出现了一处较宽的吸收峰,是因为层板上羟基中的氢键的振动引起的;在1629cm-1附近出现了一处吸收峰,这是由于结晶水中羟基的弯曲振动所引起的;在1359-1384cm-1处出现的吸收峰是因为co32-的碳氧键不对称伸缩振动;1000cm-1以下的均为ni-o、al-o、fe-o金属与氧结合的振动峰。图2与图1相似,因为钼酸铋的红外表征数据只出现在1000cm-1之后,所以不同之处就在于图2中1000cm-1以下的振动峰有所变化,807cm-1、561cm-1、494.79cm-1为bi-o、mo-o,说明钼酸铋确实存在。由图1和图2可以看出阴离子一直都是碳酸根离子。图3为本发明实施例1制备的fe3o4@ldhs的xrd谱图;图4和图5为本发明实施例1制备的fe3o4@ldhs-bi2moo6的xrd谱图和xrd放大后谱图;从图3中可以看出:fe3o4的衍射峰是(220)、(400)、(422)、(511)、(440),分别位于2θ=30.5°、44.2°、53.9°、57.1°、67.2°处;ldhs的典型特征衍射峰是(003)、(006)、(009)、(110)等,分别位于2θ=11.9°、24.6°、25.1°、61.6°处;其中(015)和(018)的峰稍有展宽,是因为水和插层阴离子的排列顺序相对较乱所导致的;由特征衍射峰(003)的2θ=12.6°和特征衍射峰(110)的2θ=64.3°可以通过计算得出晶格常数分别为c=2.44nm和a=0.37nm;再利用谢乐公式可以大致计算出晶体在c轴方向上的尺寸是28.2nm,最后由d003得到其层板间距约为0.78nm,计算后可发现每个颗粒都大约由35层层板组成。从图4中可以看出,fe3o4的衍射峰和ldhs的特征衍射峰依然存在,同时出现钼酸铋的典型特征衍射峰,钼酸铋的典型特征衍射峰是(020)、(131)、(200)、(331)等,分别位于2θ=10.6°、27.3°、31.7°、45.2°处;。只是由于钼酸铋的加入,在相对强度上有明显差别;加入了钼酸铋之后对水滑石的晶体结构产生了一定的影响,其中ldhs的典型特征峰(003)、(006)、(009)、(015)、(018)均出现减弱,而特征峰(110)、(113)增强。图6为本发明实施例1制备的fe3o4@ldhs-bi2moo6的漫反射谱图(a:bi2moo6、b:ldhs-bi2moo6、c:ldhs);从图中b曲线可以明显看出,水滑石与钼酸铋的复合材料的吸收波长向可见光方向移动,而且吸收对可见光的吸收强度有显著提高。与可见光下降解有机污染物的性能测试相符合。并且从侧面说明复合材料的成功合成。图7和图8分别为本发明实施例1制备的fe3o4@ldhs、fe3o4@ldhs-bi2moo6的sem谱图;由图7可知,fe3o4@ldhs的形貌特征可以应用扫描电子显微镜进行观察。我们可以从sem照片看出,两种物质团聚在一起,比较紧密,其中呈圆球状的为fe3o4,片状层板结构则为ldhs,部分裸露出来的层板则是未包裹上fe3o4;fe3o4@ldhs具有明显的片层状结构及六边形形状。由图8可知,我们可以从sem照片看出物质之间未团聚在一起,分散比较均匀,这是因为钼酸铋也是层板结构,大小层板叠加,钼酸铋的加入使水滑石层板分离,这说明钼酸铋已经成功与ldhs复合在一起,而且钼酸铋的加入使水滑石的层板分离与图7的水滑石层板叠加完全不同。实施例2-14步骤(2)中fe3o4、ni2+与al3+离子溶液、浓度为1m的naoh水溶液的加入量如表1所示,步骤(3)中,fe3o4@ldhs、0.4m的bi(no3)3水溶液、0.2m的na2moo4溶液水的加入量如表1所示,其余反应原料和条件类似于实施例1。表1实施例2-14中部分原料的加入量对比例1按照溶液的总离子浓度为0.6mol/l,且按照bi3+与moo4-摩尔比为2:1的要求,称取适量bi(no3)3和na2moo4·2h2o,分别放入100ml烧杯中,加入适量蒸馏水搅拌溶解之后,分别定容在100ml容量瓶中,得到bi2moo6。对比例2对比例2为空白样。试验例1光催化实验在烧瓶中加入200ml的10mg/l亚甲基蓝水溶液,放在磁力搅拌器上室温搅拌10min后取第一个样并标号为0。再放入0.1g实施例2制备得到的核壳结构光催化剂fe3o4@ldhs-bi2moo6,继续室温遮光搅拌半个小时,取第二个样并标号为1。继续室温遮光搅拌;之后每隔15min依次取一个样品,分别标号为2、3、4、5、6,一共取7个样品。将样品进行离心15min后,用紫外分光光度计检测其吸光度,利用公式可算出亚甲基蓝在不同时间内的降解效果,如表2所示。表2实施例2在遮光处理下的降解效果由表2可知,亚甲基蓝在加入核壳结构光催化剂fe3o4@ldhs-bi2moo6遮光处理30min后,显示还有82%未降解,继续在遮光条件下,随着时间的延长,基本上保持82%未降解,这是由于在无可见光情况下,核壳结构光催化剂fe3o4@ldhs-bi2moo6不能降解亚甲基蓝,初始显示出的降解,仅仅是由于亚甲基蓝被吸附在fe3o4@ldhs-bi2moo6表面,而在30min时,这种吸附基本上已达到饱和,因此,随着时间的延长,亚甲基蓝的量显示基本不变。经过大量理论研究是试验,亚甲基蓝与fe3o4@ldhs-bi2moo6在遮光处理30min后,fe3o4@ldhs-bi2moo6的吸附量基本上已达到饱和,为了明确亚甲基蓝的减少是由于在光催化剂fe3o4@ldhs-bi2moo6下的降解,而非吸附,我们在光催化实验开始之前预先遮光处理30min,使得亚甲基蓝的吸附达到饱和,之后再进行光催化,此时亚甲基蓝的减少量即可确认为全被降解。试验例2实施例2-14的光催化实验在烧瓶中加入200ml的10mg/l亚甲基蓝水溶液,放在磁力搅拌器上室温搅拌10min后取第一个样并标号为0。经过搅拌后,亚甲基蓝水溶液已混合均匀。再放入0.1g已经合成的fe3o4@ldhs-bi2moo6,继续室温遮光搅拌半个小时,取第二个样并标号为1。紧接着打开氙灯(125w。400nm滤光片)进行光催化反应;之后每隔15min依次取一个样品,分别标号为2、3、4、5、6、7、8、9、10、11,一共取11个样品。将样品进行离心15min后,用紫外分光光度计检测其吸光度,利用公式可算出亚甲基蓝在不同时间内的降解效果,如图9和表3所示。表3中,对比例1为bi2moo6,对比例2为空白样。表3实施例2-14在不同时间段的亚甲基蓝剩余量在图9和表3中,-30min处是标号为0样品中亚甲基蓝的剩余量,此时还未加入fe3o4@ldhs-bi2moo6,亚甲基蓝的剩余量为100%。0min处是标号为1的样品中亚甲基蓝的剩余量,在此之前是黑暗处理了30min,由实验例1可知,此处亚甲基蓝的减少量是由于fe3o4@ldhs-bi2moo6的吸附,再之后亚甲基蓝的减少是降解了。经过150min后,在fe3o4@ldhs-bi2moo6的吸附和降解下,亚甲基蓝可降低至28%。另外,对比例1为纯的bi2moo6,一方面成本高,另一方面单独的bi2moo6性能不稳定,无法进行回收,不能够循环利用。由于光照15min、30min等时间下的亚甲基蓝的剩余量与0min时亚甲基蓝剩余量的差值为亚甲基蓝在fe3o4@ldhs-bi2moo6光催化剂下的降解量。因此,可得出亚甲基蓝的降解量如表4所示。表4亚甲基蓝的降解量153045607590105120135150实施例20.020.070.10.140.160.260.290.360.400.44实施例30.070.140.180.240.310.370.490.570.630.7实施例40.020.080.110.170.210.270.310.340.400.45实施例50.020.060.110.160.210.270.330.400.450.5实施例60.050.090.150.210.260.310.370.440.480.51实施例70.020.070.120.180.250.310.360.410.460.52实施例80.040.080.140.200.250.300.330.340.390.45实施例90.070.140.210.220.280.380.430.490.570.64实施例100.020.060.090.130.180.220.250.290.320.35实施例110.060.110.160.230.290.350.400.440.500.53实施例120.050.090.150.200.260.320.380.420.450.48实施例130.040.070.120.160.220.260.310.350.410.45实施例140.030.070.110.160.200.260.300.330.390.45对比例10.050.090.110.150.200.270.330.370.420.46对比例20.010.010.020.030.030.040.040.050.060.07由表4可知,随着时间的延长,亚甲基蓝的降解量增加,150min时,亚甲基蓝的降解量可高达70%。试验例3fe3o4@ldhs-bi2moo6核壳结构光催化剂的循环使用对实施例3制备得到的fe3o4@ldhs-bi2moo6光催化剂做的五次循环实验,催化效率如图10所示,由图10可知,经过五次循环后,本发明制备得到的fe3o4@ldhs-bi2moo6光催化剂的催化效率几乎不变,催化效率略有降低的原因是由于催化剂的部分流失,以及催化剂上残留的上次吸附的污染物。而纯的bi2moo6光催化剂无法循环使用。综上,本发明光催化剂在可见光下光降解有机污染物具有很高的稳定性和降解率,可循环利用5次以上而活性无明显下降,降低成本。最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1