一种Pt基催化剂及其制备方法和应用与流程
本发明涉及贵金属催化剂的
技术领域:
,更具体地,涉及一种pt基催化剂及其制备方法和应用。
背景技术:
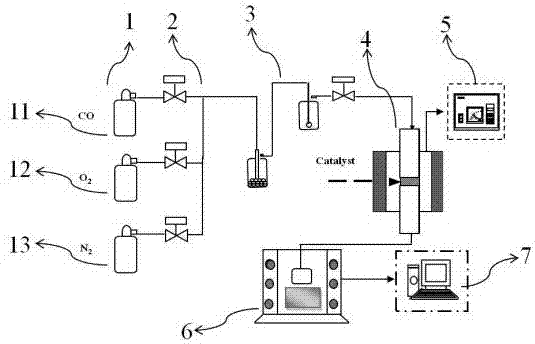
co作为煤、石油等含碳物质或烃类物质不完全燃烧的产物,也是温室气体之一,过多的排放不但会给人类身体健康带来严重威胁,同时也对生态环境带来极大的危害。对co的处理中催化氧化技术是co无害化处理中应范围最广泛,最经济有效的方法。co催化氧化反应是co与o2在催化剂表面的双分子反应,是很多工业过程中的重要反应,按照种类不同可分为贵金属催化剂、非贵金属催化剂、分子筛催化剂以及合金催化剂等,不同催化剂体系的催化氧化反应机理不同。在co催化氧化反应中,贵金属催化剂中贵金属主要包括金、银和铂族金属(铂、铑、钯、铱),贵金属催化剂价格较高。对于co催化氧化应用中的pt、au、pd等贵金属催化剂催化活性好、反应温度低,所需活性相量少,是比较常用的催化剂。催化剂制备方法不同,会影响催化剂活性组分的分散、催化剂颗粒的大小和结构等。浸渍法制备负载型贵金属催化剂的方法虽然简单,但催化剂表面活性组分分布不均,制得的负载型贵金属催化剂稳定性较差。现有的贵金属pt催化剂在高温环境下贵金属pt容易团聚失活,活性长效性差,寿命短,对贵金属pt催化剂来说并不能达到经济环保的效果。因此,需要开发出稳定性更好的用于co催化氧化的贵金属pt催化剂。技术实现要素:本发明为克服上述现有技术所述的稳定性差、在高温环境下贵金属pt容易团聚失活的缺陷,提供一种pt基催化剂的制备方法,制得的pt基催化剂能够用于co催化氧化,稳定性好,在高温环境下贵金属pt不容易团聚失活。本发明的另一目的在于提供上述制备方法制得的pt基催化剂。本发明的还一目的在于提供上述pt基催化剂在co催化氧化中的应用。为解决上述技术问题,本发明采用的技术方案是:一种pt基催化剂的制备方法,包括如下步骤:s1.将镧修饰的三氧化二铝载体la/al2o3负载pt前驱体,然后通过微波加热进行低温煅烧,得到过渡催化剂pt/la-al2o3;s2.将步骤s1.制得的过渡催化剂pt/la-al2o3和棒状体ceo2混合均匀,然后通过微波加热进行高温煅烧,得到pt基催化剂。发明人研究发现,负载pt前驱体的la/al2o3通过微波加热进行低温煅烧得到负载pt的过渡催化剂pt/la-al2o3;pt/la-al2o3与棒状体ceo2混合均匀后,通过微波加热进行高温煅烧,pt/la-al2o3上的pt被ceo2俘获,即结合微波加热和原子捕捉,成功实现pt从过渡催化剂pt/la-al2o3转移到ceo2上,形成pt/ceo2,得到pt基催化剂;并且,需要煅烧足够的时间,才能将pt/la-al2o3上的pt绝大部分转移到ceo2上。相对于pt基催化剂的传统制备方法,此时催化剂的成核方式发生改变并达到更佳稳定的状态。制得的pt基催化剂能够用于co催化氧化,稳定性好,在高温环境下贵金属pt不容易团聚失活,且催化活性明显优于通过普通加热方式或普通浸渍法制得的pt基催化剂。所述镧修饰的三氧化二铝载体la/al2o3可市售得到,也可由本领域技术人员根据现有技术制备得到。镧修饰的三氧化二铝载体la/al2o3中la的质量含量一般为3%~6%,可以选择la的质量含量为4%的la/al2o3。其中的三氧化二铝载体为本领域中常用的三氧化二铝载体。棒状体ceo2可由本领域技术人员根据现有技术制备得到。该棒状体ceo2的尺寸为(9.6±1.2)×(50~200)nm。优选地,所述pt前驱体为氯铂酸或氯铂酸铵。优选地,所述pt前驱体为氯铂酸。优选地,la/al2o3通过浸渍法负载氯铂酸。优选地,la/al2o3负载氯铂酸的方法为,将la/al2o3与氯铂酸溶液混合,然后去除溶剂。la/al2o3负载氯铂酸的具体操作:将计算量氯铂酸溶液逐滴滴加到载体la/al2o3,60℃下超声搅拌1h;浸渍2h后去除溶剂。优选地,所述低温煅烧的条件为330~370℃温度下煅烧1.0~2.0h。优选地,所述高温煅烧的条件为780~820℃温度下煅烧10~16h。负载氯铂酸的la/al2o3在较低温度330~370℃条件下煅烧1.0~2.0h得到负载pt的过渡催化剂pt/la-al2o3。pt/la-al2o3与棒状体ceo2混合均匀后,在较高温度780~820℃条件下煅烧时,pt/la-al2o3上的pt被ceo2俘获,使得pt/la-al2o3上的pt转移到ceo2上;并且需要煅烧足够的时间10~16h,才能将pt/la-al2o3上的pt绝大部分转移到ceo2上,形成pt/ceo2。优选地,步骤s2.中微波加热的功率为800~1100w。更优选地,步骤s2.中微波加热的功率为900~1000w。进一步优选地,步骤s2.中微波加热的功率为1000w。发明人研究发现,900w和1000w制得的pt基催化剂的催化活性优于800w和1100w制得的pt基催化剂,而且,其中,1000w制得的pt基催化剂的催化活性最佳。在不同的微波加热功率下,单位时间内微波源产生的微波能不同,相同的催化剂含量条件下所吸收到的微波能也有所差异。微波数值过小,加热速率慢,物质分子在微波电磁场中转动速度不够快速,会引起少量粒子的团聚现象。若功率过大,升温速率快,若升温到预设值微波煅烧炉会停止做功而导致部分微波能没被吸收。因此,选择最佳的微波功率才能达到物质内大量晶核均匀的生成以及减少粒子团聚的目的。优选地,步骤s2.中微波加热的时间为10~16h。更优选地,步骤s2.中微波加热的时间为12~16h。进一步优选地,步骤s2.中微波加热的时间为14h。发明人研究发现,co催化氧化的反应温度为90℃时,微波煅烧12h、14h和16h制得的pt基催化剂的催化活性优于微波煅烧10h制得的pt基催化剂;当co催化氧化的反应温度升至120℃时,微波煅烧14h制得的pt基催化剂的催化活性最佳。在最后一步的原子捕捉过程原理在于维持在高温环境下,将在过渡载体上以pto形式存在的pt物种气态化而被终端载体ceo2成功捕捉,因此需要较长时间高温环境的维持以保证pt颗粒的完全转移。优选地,步骤s2.中过渡催化剂pt/la-al2o3和棒状体ceo2的质量比为1.9~2.1︰1,过渡催化剂pt/la-al2o3和棒状体ceo2的混合物中pt的质量百分数为0.2%~2%。更优选地,步骤s2.中过渡催化剂pt/la-al2o3和棒状体ceo2的混合物中pt的质量百分数为1%~2%。进一步优选地,步骤s2.中过渡催化剂pt/la-al2o3和棒状体ceo2的质量比为2︰1,过渡催化剂pt/la-al2o3和棒状体ceo2的混合物中pt的质量百分数为1%。发明人研究发现,pt负载量为1%和2%的pt基催化剂的催化活性优于pt负载量为0.2%和0.6%的pt基催化剂,而且,其中,pt负载量为1%的pt基催化剂的催化活性最佳。过小的含量无法表现活性成分,含量过大又会堵塞载体的孔径而造成催化剂性能的下降。通过比表面积的测试分析可发现,当负载量为1%pt时,催化剂的比表面积及孔体积达到最大值,说明此时的含量使得活性相pt在载体ceo2上分散性最好,负载效果最佳,为最适宜的活性含量负载量。本发明同时保护上述制备方法制得的pt基催化剂。上述pt基催化剂能够用于co催化氧化,稳定性好,在高温环境下贵金属pt不容易团聚失活。本发明还保护上述pt基催化剂在一氧化碳催化氧化中的应用。优选地,进行一氧化碳催化氧化时通入的气体含有一氧化碳和氧气,所述一氧化碳的体积浓度为750~850ppm,所述氧气的体积浓度为6%~9%。进行一氧化碳催化氧化时通入的气体一般含有一氧化碳和氧气,另外通入氮气作为平衡气体。o2的存在使得o2吸附在催化剂表面形成吸附态氧,从而与进料成分中co反应,同时造成氧空位的形成,此时ceo2中的晶格氧便与co进行反应,如此循环。适宜o2的浓度促进与co的反应,但浓度过高,表明了其在催化剂表面达到饱和吸附,再增加o2浓度也不能提高催化剂的转化效率。与现有技术相比,本发明的有益效果是:本发明提供的制备方法通过结合微波加热和原子捕捉,制得含有pt/ceo2的pt基催化剂,制得的pt基催化剂能够用于co催化氧化,稳定性好,在高温环境下贵金属pt不容易团聚失活,且催化活性明显优于通过普通加热方式或普通浸渍法制得的pt基催化剂。附图说明图1为实验室模拟co催化氧化的实验室装置示意图。图中,1-钢瓶;2-质量流量控制器;3-气体缓冲瓶;4-催化剂固定床反应器;5-温度回馈控制器;6-烟道气体分析器;7-电脑-数据分析处理器。图2为实施例1制得的pt基催化剂的tem图。具体实施方式下面结合具体实施方式对本发明作进一步的说明。实施例中的原料均可可通过市售得到;除非特别说明,本发明采用的试剂、方法和设备为本
技术领域:
常规试剂、方法和设备。微波加热的设备为微波马弗炉(cy-mu1200c-s,湖南长仪微波科技有限公司)。1.镧修饰的三氧化二铝载体la/al2o3的制备将计算量的活性氧化铝al2o3倒入装有一定量镧的硝酸盐溶液的烧杯中,60℃下超声搅拌1h;浸渍2h后,去除溶剂,然后在微波煅烧炉中550w、500℃煅烧30min,得到镧修饰的三氧化二铝载体la/al2o3,其中la质量百分数为4%。本发明的实施例中使用的la/al2o3中la质量百分数均为4%。2.棒状体ceo2的制备将计算量铈的硝酸溶液(ce(no3)2·6h2o)溶于适量去离子水,均匀搅拌;将计算量氢氧化钠(naoh)溶于适量去离子水,在通风橱内进行搅拌;将上述两个溶液进行混合,并在60℃下超声搅拌1h,并将混合溶液倒入特氟龙罐,放入烘箱100℃下水热处理24h;取出特氟龙罐,待其降至室温后将混合液转移至烧杯,并用水和乙醇清洗数次,直至溶液呈中性为止,制备得到淡黄色棒状体ceo2。实施例1一种pt基催化剂的制备方法,包括如下步骤:s1.称取一定量制备好的被修饰载体4%la/al2o3装入50ml烧杯,将计算量h2ptcl6逐滴滴加其中,并于超声机中60℃下反应1h,浸渍2h后,去除溶剂,转移至石英舟并于微波煅烧炉中450w、350℃下煅烧2h,得到过渡催化剂pt/la-al2o3;s2.将步骤s1.中pt/la-al2o3与制备好的淡黄色棒状体ceo2按2:1的质量比进行物理混合,在研钵中均匀研磨15min,其中,保证pt的质量分数为1%;将研磨好的样品于微波煅烧炉中1000w下煅烧14h,制得pt基催化剂。实施例2~4与实施例1不同的是,实施例2~4中步骤s2.的煅烧功率分别为800w、900w和1100w,如表1所示;其他原料使用量和操作步骤与实施例1相同。实施例5~7与实施例1不同的是,实施例5~7中步骤s2.的煅烧时间分别为10h、12h和16h,如表1所示;其他原料使用量和操作步骤与实施例1相同。实施例8~10与实施例1不同的是,实施例8~10中步骤s2.的混合物中pt负载量分别为0.2%、0.6%和2%,如表1所示;其他原料使用量和操作步骤与实施例1相同。表1实施例2~10的pt基催化剂的制备条件对比例1与实施例1不同的是,本对比例中步骤s1.和s2.的煅烧均采用普通马弗炉煅烧;其他原料使用量和操作步骤与实施例1相同。性能测试1.透射电镜测试透射电子显微镜(transmissionelectronmicroscope(tem),oxfordincaenergy400);高分辨率下获取全貌图,选择不同载体ceo2、la-al2o3进行能谱的扫描(eds);以验证pt的转移成功性。2.催化剂的co催化氧化活性测试(1)催化剂的co催化氧化过程:催化剂在含氧状态下与co反应方程式如下:o2+co*→co2+o*①o*+co→co2②co优先与吸附态的o2反应生成co2,此后催化剂上的晶格氧能迅速补充此过程产生的氧缺位继续反应,如此循环。(2)实验室装置:实验室装置图如图1所示;实验室模拟co催化氧化反应操作表示为:先由气体系统调配所需的反应气体,根据计算所需进料成分,钢瓶出来后的气体通过质量流量计调节浓度和流量。配制好的气体在气体混合瓶混合后均匀进入配有温度调节系统的催化剂反应装置进行活性测试,连接烟气分析仪进行反应前后的气体浓度分析,随后经过如下方程式③,计算出co转化率,比较催化剂的活性。方程式③中,cco,in表示co的进口浓度;cco,out表示co的出口浓度。(3)测试方法:首先先称取0.03g石英棉花于直径为10mm、管长为80cm的石英反应管中心十子形支架上,作为支撑催化剂粉末的隔热挡板;取催化剂0.22g放入该石英反应管中,反应前先通入含有5%h2的混合气体在800℃下处理1h,空气中500℃反应30min,后冷却至室温再通入反应气体800ppmco+9%o2+n2(n2为平衡气体),总流速为500ml/min,空间速度ghsv=100ⅹ10^3h-1,反应温度为60~300℃,程序升温,并依次在60℃、90℃、120℃、150℃、180℃、210℃、240℃、270℃和300℃时稳定一段时间,记录该温度下co转化率。烟气分析仪(pg-350)在线实时监测,电脑处理数据。3.催化剂的co催化氧化稳定性测试取催化剂0.22g于石英反应管中进行上述活性测试的步骤。进行一次程序升温活性测试后关闭气瓶,等加热电炉冷却至室温,并通入n2除去表面杂质。随后,再次通入反应气体进行下一次程序升温活性测试,方法同上。总共进行5次活性测试,依次记作第1次、第2次、第3次、第4次及第5次。反应完毕后,计算催化剂进行的5次测试下co转化率,以此比较不同催化剂的稳定性。本发明的实施方式中,比较了实施例1与对比例1制得的pt基催化剂的稳定性。测试结果tem测试结果如图2所示,呈块状的为过渡载体la/al2o3,棒条状的为载体ceo2,从图中可观察到一些pt颗粒附着在棒状ceo2载体上。进一步结合能谱的扫描,分别选取图上la/al2o3及ceo2部分进行能谱扫描,能谱扫描结果如表2所示,可见绝大部分pt金属已从过渡载体la/al2o3上移动并被棒状体ceo2载体成功捕捉,验证了pt基催化剂中pt/ceo2的成功制备。表2实施例1制得pt基催化剂的tem能谱扫描结果(归一化质量百分比/%)laaloceptla/al2o33.2947.2243.366.030.1ceo200.0438.5658.203.20表3实施例1制得的pt基催化剂的稳定性测试结果表4对比例1制得的pt基催化剂的稳定性测试结果根据表3和表4可知,co催化氧化的反应温度为120℃时,测试五次后,实施例1制得的pt基催化剂的co转化率从95%仅仅下降至88%,co转化率变化较小,能够保持良好的催化活性;而对比例1制得的pt基催化剂的co转化率则从48%大幅下降至5%,co转化率变化巨大。可见,实施例1制得的pt基催化剂不仅催化活性显著优于对比例1制得的pt基催化剂,而且稳定性也显著优于对比例1制得的pt基催化剂。相对于普通加热方式制得的pt基催化剂,实施例1中采用微波加热结合原子捕捉的方法制得的pt基催化剂能够保持良好的催化活性,在高温环境下贵金属pt不容易团聚失活。另外,发明人通过改变微波功率、微波煅烧时间和pt负载量,研究不同制备参数对pt基催化剂的催化活性的影响。发明人研究发现,将实施例1~4中采用不同功率制得的pt基催化剂用于co催化氧化,co催化氧化的反应温度为90℃时,900w和1000w制得的pt基催化剂的co转化率分别为29%和28%,优于800w和1100w制得的pt基催化剂,800w和1100w制得的pt基催化剂的co转化率分别为20%和6%。反应温度为120℃时,800w、900w、1000w和1100w制得的pt基催化剂的co转化率分别为66%、65%、90%和61%,可见1000w制得的pt基催化剂催化活性最佳。将实施例1及实施例5~7制得的pt基催化剂用于co催化氧化,co催化氧化的反应温度为90℃时,微波煅烧10h制得的pt基催化剂的co转化率仅仅为12%,而微波煅烧12h、14h和16h制得的pt基催化剂的co转化率分别为30%、28%和31%,可见实施例1及实施例6~7制得的pt基催化剂的催化活性优于实施例5。co催化氧化的反应温度为120℃时,微波煅烧14h制得的pt基催化剂的co转化率能够达到90%,而微波煅烧12h和16h制得的pt基催化剂的co转化率分别为67%和66%,可见微波煅烧14h制得的pt基催化剂的活性最佳。将实施例1及实施例8~10制得的pt基催化剂用于co催化氧化,co催化氧化的反应温度为90℃时,pt负载量为1%和2%的pt基催化剂的co转化率分别为28%和30%,而pt负载量为0.2%和0.6%的pt基催化剂的co转化率仅仅分别为1%和18%,可见实施例1和实施例10制得的pt基催化剂的催化活性优于实施例8~9。co催化氧化的反应温度为120℃时,pt负载量为1%和2%的pt基催化剂的co转化率分别为90%和63%,而pt负载量为0.2%和0.6%的pt基催化剂的co转化率仅仅分别为9%和46%,所以,pt负载量为1%的pt基催化剂的催化活性最佳。此外,发明人还研究了不同氧气浓度对本发明的pt基催化剂催化氧化co的影响。设置氧气浓度分别为0、3%、6%、9%和12%,其他测试条件不变,测试实施例1制得的pt基催化剂的co转化率。co催化氧化的反应温度为90℃时,氧气浓度9%时pt基催化剂催化co的转化率为90%,无氧条件下的co转化率为0,而氧气浓度分别为3%、6%和12%时pt基催化剂催化co的转化率分别为16%、55%和39%。可见,进料气体成分中9%o2浓度是促进co与催化剂上晶格氧接触反应的最佳浓度。显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1