茜素红-S螯合形成树脂的合成方法及其应用与流程
本发明属于贵金属分离富集用吸附材料技术领域,具体涉及一种螯合形成树脂的合成方法及其应用。
背景技术:
贵金属元素在地壳中含量较低,常与cu、ni、zn、mo、pb等贱金属元素伴生。由于贵金属元素具有较强的颗粒效应及延展性,为了使样品具有代表性,在分析测试时必须加大样品的取样量以克服颗粒效应及延展性的影响,因此,在溶解贵金属的同时,大量的贱金属离子也同时被溶解,待测溶液中cu、ni、cd、mo、pb等离子严重干扰icp-ms对贵金属元素测定。
贵金属元素的分离富集,不仅能消除共存离子的质谱干扰,而且能有效降低icp-ms测定的基体效应。因此,贵金属富集剂的研究成为越来越多的贵金属分析工作者关注的焦点。报道应用最多的是在盐酸介质中,贵金属离子与溶液中cl-生成络阴离子,通过717阴离子交换树脂与活性炭相结合的吸附方式可以实现au、pt、pd与大部分贱金属离子的分离。另外,由于贵金属元素同属过渡元素,其d轨道中的电子均未充满,易与能够给出电子的官能团(含有硫、氮、氧等原子)结合形成螯合物,使贵金属元素被螯合树脂选择性吸附,从而达到与贱金属离子分离的目的。但这两种分离富集剂均对介质要求较高,而且通常需要与活性炭联合使用,才能满足au、pt、pd同时吸附并达到理想的效果。
螯合形成树脂(国外称为万能树脂)是目前更为优良的吸附剂,其吸附选择性较高,吸附容量相对较大,且能够在酸、碱介质中稳定存在,可以在较宽的温度和酸度范围内选择性的吸附贵金属元素。本文采用1,2-二羟基蒽醌-3-磺酸钠(茜素红-s)作为螯合剂,与大孔强碱性阴离子树脂d296r合成螯合形成树脂,并对吸附性能进行了研究,发现其对au、pt、pd具有较好的吸附选择性。
技术实现要素:
针对现有技术中存在的问题,本发明提供一种螯合形成树脂的合成方法及其应用,具有吸附选择性高,吸附容量较大,且能够在酸、碱介质中稳定存在的特性,可以在较宽的温度和酸度范围内选择性的吸附贵金属元素。
为解决上述技术问题,本发明采用以下技术方案:
一种茜素红-s螯合形成树脂的合成方法,包括以下步骤:
(1)将1,2-二羟基蒽醌-3-磺酸钠加入去离子水中,完全溶解后制得浓度为10g/l的1,2-二羟基蒽醌-3-磺酸钠水溶液,备用;
(2)称取50g预处理后的d296-r型大孔强碱性阴离子交换树脂于250ml烧杯中,用4mol/l的hcl溶液浸泡24小时以上,使其充分溶胀转型后,用去离子水洗至排出水为中性;
(3)向经步骤(2)处理后的d296-r型大孔强碱性阴离子交换树脂中加入50ml浓度为10g/l的1,2-二羟基蒽醌-3-磺酸钠水溶液,不断搅拌,9-12min后,弃去上层清夜,继续加入50ml浓度为10g/l的1,2-二羟基蒽醌-3-磺酸钠水溶液,反复处理,直至上层溶液不再变清为止;弃去溶液,用去离子水洗至不再脱色,即得到茜素红-s螯合形成树脂。
所述步骤(2)中d296-r型大孔强碱性阴离子交换树脂的预处理方法如下:
将d296-r型大孔强碱性阴离子交换树脂装入烧杯中,用2倍树脂体积的浓度为10wt%nacl溶液浸泡18-20小时,弃去食盐水,用去离子水漂洗,直至洗水不呈黄色为止;用2倍树脂体积的体积分数为5%盐酸浸泡2-4小时,弃去酸溶液,用去离子水冲洗树脂至排出水为中性,除去铁、铝、钙、镁等无机杂质;用2倍树脂体积的浓度为(2-4)wt%的氢氧化钠溶液浸泡2-4小时,弃去碱溶液后,用去离子水冲洗树脂至排出水为中性,可除去有机物和硅等无机杂质;经上述处理后,树脂已变成oh型,经低温风干后,称取树脂各200g,粉碎后过80目筛,备用。
所述的茜素红-s螯合形成树脂在贵金属分析中的应用。
所述的茜素红-s螯合形成树脂的应用,步骤如下:
(1)吸附装置的制作
a、定量滤纸纸浆:称取100g中速定量滤纸加热水搅拌揉碎,补加水至10l,即得10g/l的纸浆,使用时稀释成5g/l;
b、吸附柱的制作:将1,2-二羟基蒽醌-3-磺酸钠螯合形成树脂溶于去离子水中得到浓度为50g/l的螯合形成树脂悬浊液,向吸附柱(φ32mm)中加入15ml步骤a制得的5g/l定量滤纸纸浆并压紧,向600ml5g/l纸浆中加30ml50g/l的螯合形成树脂悬浊液,搅匀后每次15ml分两次分别将其加入20个吸附柱,压紧,再次加入15ml5g/l定量滤纸纸浆后压紧,减压抽虑,上置布氏漏斗;
(2)样品处理及检测
1)准确称取10.0g(精确至0.01g)试样于瓷舟中,将瓷舟置于低温马弗炉中,升温到650℃~700℃灼烧2h,取出冷却,将试样转入200ml聚四氟乙烯高压密闭溶样罐中,加水润湿,加入30mlhcl、5mlh2o2、2gkclo3,或30ml新配制浓王水(30mlhcl、5mlh2o2、2gkclo3和30ml新配制浓王水两种溶解体系在溶解过程中,与试样发生反应并有少量挥发,在样品完全溶解并打开溶样罐时,预计剩余15~20mlhcl或王水,再加入约150ml热水,倒入制作好的吸附柱上的布氏漏斗中,此时分离富集的酸度约为10%的hcl或王水介质),混匀,将聚四氟乙烯溶样罐用高压密闭罐压紧,置于恒温鼓风干燥箱升温至100℃保温1h,再升温至160℃消解5h,取出高压密闭溶样罐冷却至室温,打开;
2)加入0.6g/lki溶液3ml搅拌充分反应后,再加入约150ml热水,搅匀,将试液及酸不溶物倒入制作好的吸附柱上的布氏漏斗中,滤速控制为l0~15ml/min,减压抽滤,取出纸饼,将其放入15ml瓷坩埚中,置于低温马弗炉内,升温到700℃至灰化完全;取出瓷坩埚,冷却后滴加2滴水润湿,再加入5ml新配制的浓王水置于电热板上加热溶解,移入10ml比色管中,用水稀释至刻度,摇匀;icp-ms测定,随同试样做空白试验。
所述步骤2)减压抽滤,抽干后,用5%(v/v)王水洗液洗涤聚四氟乙烯溶样罐及布氏漏斗各3次,取下布氏漏斗,用热的20g/lnh4hf2溶液及热的5%(v/v)hcl分别洗涤吸附柱3次,每次10ml,再用热水洗涤吸附柱,每次10ml,洗涤2次,抽干。
茜素红-s螯合形成树脂的合成及吸附机理:按照树脂预处理方法充分洗涤d296-r型强碱性阴离子交换树脂后,风干、粉碎,浸泡于4mol/l的盐酸介质中,d296-r型强碱性阴离子交换树脂上的-oh与-cl完全交换,使r-oh型的d296-r树脂转变成r-cl型树脂(见式1)。配制的螯合剂茜素红-s(1,2-二羟基蒽醌-3-磺酸钠)溶液在水中离解出na+后,变成一个较大的阴离子(见式2),此阴离子与r-cl型树脂相遇发生离子交换,取代了r-cl型树脂上的氯离子,茜素红-s被稳定的吸附在d296-r型强碱性阴离子交换树脂上,即合成了茜素红-s螯合形成树脂(见式3)。在盐酸介质中,茜素红-s螯合形成树脂中的-oh离解出h+,变成-o-(见式4),此时,贵金属元素以氯络阴离子形式存在,与茜素红-s螯合形成树脂相遇,贵金属离子即被吸附在树脂上而形成螯合物,以pd为例,作用机理见式5。
本发明的有益效果:本发明的茜素红-s螯合形成树脂与717阴离子交换树脂及一般的螯合树脂相比,具有吸附选择性高,吸附容量较大,且能够在酸、碱介质中稳定存在的特性,可以在较宽的温度和酸度范围内选择性的吸附贵金属元素。将其与高压密闭分解样品相结合,有效分离贱金属离子,提高了au、pt、pd的回收率,成本低廉、分析效率高,节约了成本。适用于大批化探样品分析,使用本发明的茜素红-s螯合形成树脂完成铂族元素地球化学成分分析一级标准物质中au、pt、pd的分离富集,回收率均大于92%,精密度、正确度满足相关质量规范要求。同时避免了处理、改进活性炭的繁琐手续,与高压密闭溶矿方式相结合,提高了分析效率,节约了成本。
附图说明
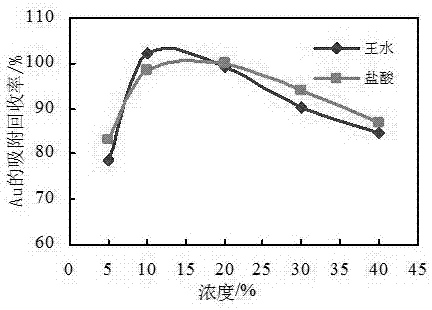
图1为螯合形成树脂对au的吸附率;
图2为螯合形成树脂对pt的吸附率;
图3为螯合形成树脂对pd的吸附率。
具体实施方式
下面结合具体实施例,对本发明做进一步说明。应理解,以下实施例仅用于说明本发明而非用于限制本发明的范围,该领域的技术熟练人员可以根据上述发明的内容作出一些非本质的改进和调整。
主要试剂和标准溶液
au、pt、pd待测元素标准溶液:au、pt、pd的标准储备液均采用光谱纯或纯度大于99.99%的金属配制成1.000mg/ml的标准储备液。将含有au、pt、pd的标准储备溶液逐级稀释配制成ρ(au、pt、pd)=1.00μg/ml的混合标准工作溶液,
分别移取ρ(au、pt、pd)=1.00μg/ml的标准工作溶液0.00、0.10、0.20、0.50、1.0、2.5、5.0、10.0、25.0ml于100ml容量瓶中,用10%(体积分数)hno3稀释至刻度、摇匀,标准系列中au、pt、pd的质量浓度依次为:0.00、1.00、2.00、5.00、10.0、25.0、50.0、100.0、250.0ng/ml。
1,2-二羟基蒽醌-3-磺酸钠(茜素红-s):国药集团化学试剂有限公司
ki(分析纯):配制0.6g/l的ki溶液。
质谱最佳调谐液(国家标准物质研究中心):2ng/ml的li、co、in、u标准溶液。
内标溶液:ρ(lu)=10ng/ml,介质为3%(体积分数)hno3,测试时由微型三通在线加入。
hcl、hno3为优级纯。
实验用水均为超纯水机制得的超纯水(电阻率为18mω·cm)。
氩气为高级纯(氩质量分数大于99.99%)。
d296-r型大孔强碱性阴离子交换树脂(南开大学化工厂)。
实施例1螯合形成树脂的合成
(1)称取10g1,2-二羟基蒽醌-3-磺酸钠(茜素红-s)粉末于500ml烧杯中,加入去离子水不断搅拌,直至粉末完全溶解,定容至1000ml容量瓶中,即得10g/l的1,2-二羟基蒽醌-3-磺酸钠水溶液,备用;
(2)d296-r型大孔强碱性阴离子交换树脂的预处理方法:将树脂装入烧杯中,用约2倍树脂体积的10%nacl溶液浸泡18-20小时,弃去食盐水,用去离子水漂洗,直至洗水不呈黄色为止;用约2倍树脂体积的5%盐酸浸泡2-4小时,弃去酸溶液,用去离子水冲洗树脂至排出水为中性,可除去铁、铝、钙、镁等无机杂质;用约2倍树脂体积的2-4%的稀氢氧化钠溶液浸泡2-4小时,弃去碱溶液后,用去离子水冲洗树脂至排出水为中性,可除去有机物和硅等无机杂质。经上述处理后,树脂已变成oh型,经低温风干后,称取树脂各200g,粉碎后过80目筛,备用;
(3)称取50g预处理后的d296-r型大孔强碱性阴离子交换树脂于250ml烧杯中,用4mol/l的hc1溶液浸泡24小时以上,使其充分溶胀转型后,去离子水洗至排出水为中性。向树脂中加入约50ml10g/l的1,2-二羟基蒽醌-3-磺酸钠水溶液,不断搅拌,约十分钟后,弃去上层清夜,继续加入50ml10g/l的1,2-二羟基蒽醌-3-磺酸钠水溶液,反复处理,直至上层溶液不再变清为止。弃去溶液,用去离子水洗至不再脱色,即得到1,2-二羟基蒽醌-3-磺酸钠(茜素红-s)螯合形成树脂。
实施例2螯合形成树脂的应用
(1)吸附装置的制作
a、定量滤纸纸浆:称取100g中速定量滤纸加热水搅拌揉碎,补加水至10l,即得10g/l的纸浆,使用时稀释成5g/l。
b、吸附柱的制作:向吸附柱(φ32mm)中加入15ml5g/l定量滤纸纸浆并压紧。向600ml5g/l纸浆中加入处理好的30ml50g/l的螯合形成树脂悬浊液,搅匀后每次15ml分两次分别将其加入20个吸附柱,压紧,再次加入15ml5g/l定量滤纸纸浆后压紧,减压抽虑,上置布式漏斗。
(2)样品处理及检测
1)准确称取10.0g(精确至0.01g)试样于瓷舟中,将瓷舟置于低温马弗炉中,升温到650℃~700℃灼烧2h。取出冷却,将试样转入200ml聚四氟乙烯高压密闭溶样罐中,加水润湿,加入30mlhcl、5mlh2o2、2gkclo3,或30ml新配制浓王水,混匀,将聚四氟乙烯溶样罐用高压密闭罐压紧,置于恒温鼓风干燥箱升温至100℃保温1h,再升温至160℃消解5h。取出高压密闭溶样罐冷却至室温,打开。
2)加入0.6g/lki溶液3ml搅拌充分反应后,再加入热水约150ml,搅匀,将试液及酸不溶物倒入制作好的吸附柱上的布氏漏斗中,滤速控制为l0~15ml/min,减压抽滤。抽干后,用5%(v/v)王水洗液洗涤聚四氟乙烯溶样罐及布氏漏斗各3次,取下布氏漏斗。用热的20g/lnh4hf2溶液及热的5%(v/v)hc1分别洗涤吸附柱3次,每次10ml,再用热水洗涤吸附柱,每次10ml,洗涤2次,抽干。取出纸饼,将其放入15ml瓷坩埚中,置于低温马弗炉内,升温到700℃至灰化完全。取出瓷坩埚,冷却后滴加2滴水润湿,再加入5ml浓王水(新配制)置于电热板上加热溶解,移入10ml比色管中,用水稀释至刻度,摇匀。icp-ms测定,随同试样做空白试验。
对比实验
d296-r型强碱性阴离子交换树脂与茜素红-s螯合形成树脂对贵金属元素的吸附
分取1mlρ(au、pt、pd)=1.00μg/ml的混合标准溶液至250ml锥形瓶中,加入50ml10%(v/v)盐酸溶液,分别将溶液倒入按上述步骤b制备吸附柱方法制备好的不同量的d296-r型大孔强碱性阴离子交换树脂吸附柱和茜素红-s螯合形成树脂吸附柱中,减压抽虑。取出滤饼放于15ml瓷坩埚中,置于马弗炉由低温升至700℃灰化2h,冷却后,用5ml新配制的王水在约为160℃恒温电热板上加热溶解,待溶解完全且无气泡产生,取下冷却,用去离子水转移至10ml比色管中,定容至刻度,摇匀,icp-ms测定。结果见表1。
表1d296-r型强碱性阴离子交换树脂和茜素红-s螯合形成树脂对照结果
从实验数据可知,改性前的d296-r型大孔强碱性阴离子交换树脂对au的吸附回收率基本可以达到80%以上,对pt、pd的吸附率却仅为50%~60%,而合成的茜素红-s螯合形成树脂对au、pt、pd的吸附率明显提高,尤其对pt、pd的吸附率提高的程度基本都大于50%。从表3还可以看出,合成的茜素红-s螯合形成树脂用量对au、pt、pd的吸附率没有太大影响,从分析成本及灰化时间考虑,选择每个样品茜素红-s螯合形成树脂的用量约为0.075g。
一、ki用量对茜素红-s螯合形成树脂吸附贵金属元素的影响
在hcl介质中,贵金属离子以[mcl4]2-的络阴离子形式存在,对贵金属有吸附作用的阴离子交换树脂及螯合树脂通常与此类贵金属络阴离子交换配位。通过试验发现加入ki溶液后,[mcl4]2-转变为体积较大的络阴离子[mi4]2-,更有利于交换配位反应的进行。因此,将ki配制成0.6g/lki溶液,分别在100ml含1000ng的au、pt、pd混合标准溶液加入不同量的ki溶液,充分反应,用研制的茜素红-s螯合形成树脂对[mi4]2-络阴离子进行配位吸附,考察ki的用量对[mcl4]2-络阴离子转换为[mi4]2-络阴离子的影响及螯合形成树脂对au、pt、pd的吸附能力,结果见表2。
表2ki用量对贵金属络阴离子交换配位影响实验结果
实验发现加入少许0.6g/lki溶液后,树脂对au、pt、pd的吸附回收率有了明显的提高,当ki溶液的加入量为3ml时,螯合形成树脂对au、pt、pd的吸附率均能达到90%以上,也就是说,0.6g/lki溶液3ml基本能保证[mcl4]2-络阴离子转换为[mi4]2-络阴离子,因此,试验确定0.6g/lki溶液为3ml。
二、吸附介质及酸度的选择性试验
取1mlρ(au、pt、pd)=1.00μg/ml的混合标准溶液若干份至250ml锥形瓶,分别配制为5%、10%、20%、30%、40%(v/v)的盐酸和王水介质,按动态吸附试验,考察研制的茜素红-s螯合形成树脂对au、pt、pd吸附效果,结果见图1~图3。由图可知,茜素红-s螯合形成树脂对au、pt、pd的吸附基本不受介质的影响,而且在10%~20%的盐酸或王水介质中,螯合形成树脂对au、pt、pd达到较为理想的吸附状态。在实际样品分析中通常选择体积分数为的10%的盐酸或王水介质。
三、金属离子的干扰
贵金属矿物通常与贱金属cu、ni、zn、mo、pb等元素伴生,采用hcl-h2o2-kclo3或王水高压密闭溶解样品时,大量cu、ni、zn、mo、pb等贱金属也被溶解,因此,必须通过分离富集,消除共存离子的干扰。由于贵金属元素同属过渡元素,具有d轨道未被充满的电子层结构,其原子轨道与配位体(如f-、cl-、br-、i-等)的原子轨道通过杂化或掺和作用而形成分子轨道,从而生成稳定的络合物。此络阴离子易与能够给出电子的官能团(含有硫、氮、氧等原子)结合形成螯合物,使贵金属元素被螯合形成树脂选择性吸附,从而达到与贱金属离子分离的目的。
试验通过向贵金属标准溶液中加入100倍量的cu、ni、zn、mo、pb贱金属离子,经螯合形成树脂吸附分离,icp-ms分别测定滤液中和螯合形成树脂灰化、王水提取后溶液中的cu、ni、zn、mo、pb贱金属元素和贵金属元素。实验结果显示,滤液中依然存在大量cu、ni、zn、mo、pb离子,螯合形成树脂吸附吸附的少量cu、ni、zn、mo、pb离子对icp-ms测定au、pt、pd没有明显的干扰。
四、方法的线性方程和检出限
在icp-ms最佳的仪器条件下测定贵金属元素au、pt、pd混合标准工作溶液,所测元素在给定范围内标准曲线的线性关系较好,其线性方程(其中y表示待测元素的质量浓度,x表示其对应的信号强度)和相关系数见表4。按照样品分析方法对全流程空白溶液连续12次测定,以测定结果的3倍标准偏差为方法的检出限,所得结果见表3。
表3线性范围、线性方程、相关系数和检出限
五、实际样品分析
称取铂族元素地球化学成分分析标准物质gbw07289、gbw07291、gbw07293、gbw07340若干份,按样品处理及检测方法,分别采用王水、盐酸-氯酸钾-过氧化氢二种高压密闭溶解方式分解样品,平行测定6次,考察研制的茜素红-s螯合形成树脂对实际样品的吸附效果,结果见表4。
表4螯合形成树脂对地质样品中au、pt、pd的吸附
由表4可知,研制的茜素红-s螯合形成树脂在不同的介质中能够很好的完成au、pt、pd的分离富集,标准物质测定结果与认定值的相对误差(re)为-7.58%~10.9%,相对标准偏差(rsd)均小于9.86%。
本发明所用仪器及工作条件
x-seriesⅱ型电感耦合等离子体质谱仪(美国thermo公司),仪器工作参数见表5。
表5等离子体质谱仪工作参数
hy-8a数显调速多用振荡器(金坛市顺华仪器有限公司)。
gzx-9240mbz数显鼓风干燥箱(上海博讯实业有限公司)。
xj系列高压密闭溶样罐(滨海县正红塑料仪器厂):容量为200ml。
吸附柱:40ml。
布氏漏斗:φ80mm。
以上显示和描述了本发明的基本原理和主要特征以及本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
- 还没有人留言评论。精彩留言会获得点赞!