一种S型异质结催化剂及其制备方法、应用
一种s型异质结催化剂及其制备方法、应用
技术领域
1.本发明属于光催化剂技术领域,具体涉及一种s型异质结催化剂及其制备方法、应用。
背景技术:
2.自工业革命以来的,对煤、石油、天然气等化石燃料的过度依赖,导致co2大量排放,大气中的co2浓度逐年升高,是全球气候变化的主要因素。然而随着全球人口的持续增长,化石燃料的使用量将继续增加,在目前阶段,世界能源需求的85%以上是由化石燃料燃烧提供的。尽管新能源技术正在逐步推进,但其增长速度并不能满足全球人口不断增长的能源需求,在未来几十年内化石能源仍旧是人类主要依赖的能源模式,co2排放放缓的可能性很小。因此,促进大气中co2的捕集和封存以及资源化利用是缓解温室效应的核心战略,因为它既满足了中短期内对化石能源日益增长的需求,同时又能减少相关的温室气体排放。在实际工业过程中能够利用co2的反应并不多,只有加氢还原、催化重整等,并且这些过程往往需要高温、高压等较为苛刻的反应条件,是高能耗、低效率的过程。从资源、能源发展战略的角度来看,利用清洁可再生的太阳能将co2高效还原成化学品或燃料,能够变废为宝,有利于人为实现碳循环的闭环,有助于可持续发展,对于缓解能源与环境双重压力具有重要的现实意义。在众多还原产物中,co是c1化学的基础,作为合成气和各类煤气的主要组分,co是合成一系列基本有机化学品和中间体的重要原料,由co出发,可以制取几乎所有的基础化学品。
3.具有二维层状结构的层状双金属氢氧化物(ldhs)因其具有可见光响应、较大的比表面积和较短的载流子扩散路径而受到广泛关注。但是,为了满足氧化还原反应发生的条件,材料需要有较大的禁带宽度;然而为了提高光响应范围,材料需要具有更小的禁带宽度,较强的氧化还原能力和较宽的光响应范围之间缺乏良好的相容性。此外,单组分催化剂受光激发产生的电子-空穴对在强库仑力作用下很容易发生重组,类似一个人在地球上跳跃时收到的引力一样,电子与空穴复合后以热量、荧光等形式耗散,大大限制了材料的活性。因此,作为典型的还原型光催化剂,如何进一步提高ldhs的光捕获能力和电荷分离能力,提高催化剂活性仍然是一个巨大的挑战。而异质结工程能够将不同能级的组分集成在一起,不同组分的界面电荷极化和内部电势梯度能够调节异质结表面电荷状态,促进电荷的分离和转移。
4.目前,ldhs基异质结用于高效co2光催化转化的报道非常少,催化效果也不尽人意。因此,构建s型ldhs基异质结是一个提升co2还原活性的潜在策略。
技术实现要素:
5.本发明的目的在于,针对现有技术的不足,提供了一种由niin ldh和in2s3组成的s型异质结光催化剂,以mil-68(in)为自牺牲模板合成了棱柱状2d交联纳米片组成niin ldh,然后通过一步硫化的方式在niin ldh纳米片上外延生长in2s3,2d纳米片组成的棱柱状
n型相比,co生成率大大提高。
附图说明
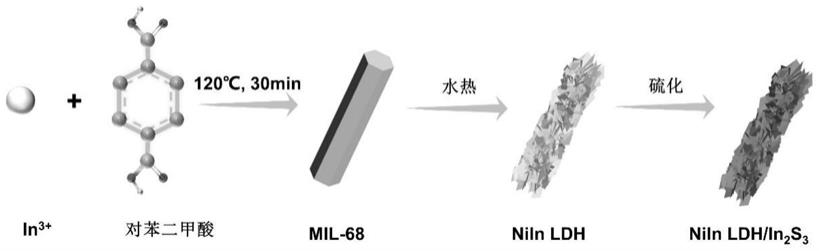
24.图1为本发明中实施例1-3制备的工艺流程图;
25.图2为本发明中实施例1-3制备的催化剂和对比例1的催化剂的扫描显微镜图;
26.图3为本发明中实施例1-3制备的催化剂和对比例1的催化剂的x射线衍射图;
27.图4为本发明中实施例2-3制备的催化剂和对比例1的催化剂的拉曼光谱图;
28.图5为本发明中实施例2-3制备的催化剂和对比例1的x射线光电子能谱谱图;
29.图6为本发明中实施例2制备的催化剂的氮气吸-脱附等温线;
30.图7为本发明中实施例2制备的催化剂的氮气吸-脱附等温线;
31.图8为本发明中实施例2制备的催化剂的氮气吸-脱附等温线;
32.图9为本发明中实施例2-3制备的催化剂和对比例1的催化剂的co2吸-脱附等温线;
33.图10为本发明中实施例2-3制备的催化剂和对比例1的催化剂的co2吸-脱附等温线;
34.图11为本发明中实施例2制备的催化剂和对比例1的催化剂的莫特肖特曲线;
35.图12为本发明中实施例2-3制备的催化剂和对比例1的催化剂的光致发光光谱和时间分辨性光致发光光谱;
36.图13为本发明中实施例2-3制备的催化剂和对比例1的催化剂的瞬态光电流响应谱;
37.图14为本发明中实施例3制备的催化剂的co与ch4产量随时间变化关系;
38.图15为本发明中实施例2-3和对比例1的制备的催化剂的co产量对比图;
39.图16为本发明中实施例2-3和对比例1的制备的催化剂及实施例2和对比例1催化剂共混co与ch4产量随时间变化关系;
40.图17为本发明中实施例3制备的催化剂与其它催化剂的co产率对比图;
41.图18为本发明中实施例3制备的催化剂在不同条件下的活性对比图;
42.图19为本发明中实施例3制备的催化剂的co2还原循环试验;
43.图20为本发明中实施例2和对比例1制备的催化剂的功函数、费米能级计算结果图;
44.图21为本发明中实施例3制备的niin ldh/in2s3的态密度计算结果图;
45.图22为本发明中实施例3制备的niin ldh/in2s3的原位拉曼光谱图;
46.图23为本发明中实施例3制备的niin ldh/in2s3催化剂能带结构与电子转移模型、机理示意图;
47.图24为本发明中实施例2-3制备的催化剂和对比例1制备的催化剂的光催化co2还原步骤自由能计算结果。
具体实施方式
48.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。基于本
发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
49.需要说明的是,本发明中所使用的专业术语只是为了描述具体实施例的目的,并不是旨在限制本发明的保护范围,除非另有特别说明,本发明以下各实施例中用到的各种原料、试剂、仪器和设备均可通过市场购买得到或者通过现有方法制备得到。
50.实施例1
51.合成mil-68(in)催化剂
52.取60mg in(no3)3·
xh2o和60mg对苯二甲酸溶于40mln,n-二甲基甲酰胺溶液,剧烈搅拌5min;然后,将溶液置于120℃油浴锅中保持30min,通过真空抽滤,并用去离子水和乙醇分别洗3次得到白色沉淀;60℃真空干燥24h,即得到mil-68(in)。
53.实施例2
54.合成niin ldh催化剂
55.取120mgs1得到的mil-68(in)分散于20ml 0.1m naoh溶液中得到混合溶液a,360mg ni(no3)2·
6h2o溶于20ml去离子水得到混合溶液b,将混合溶液a和混合溶液b充分混合,转移到50ml聚四氟乙烯内胆不锈钢反应釜中,180℃下保持反应12h;冷却至室温后,真空抽滤,并用去离子水和乙醇分别洗涤3次得到绿色沉淀;60℃真空干燥24h,得到niin ldh。
56.实施例3
57.合成s型异质结niin ldh/in2s3催化剂
58.取60mg得到的niin ldh分散于40ml乙醇,在剧烈搅拌下加入60mg硫脲,继续剧烈搅拌30min后,转移至50ml聚四氟乙烯内胆不锈钢反应釜,100℃下保持反应2h,冷却至室温后,真空抽滤,并用去离子水和乙醇分别洗涤3次得到灰色沉淀;60℃真空干燥24h,得到niin ldh/in2s3。其反应原理示意图如图1所示。
59.实施例4
60.一种s型异质结催化剂的制备方法,包括如下步骤:
61.s1、合成mil-68(in):取60mg in(no3)3·
xh2o和60mg对苯二甲酸溶于40mln,n-二甲基甲酰胺溶液,剧烈搅拌3min;然后,将溶液置于120℃油浴锅中保持30min,通过真空抽滤,并用去离子水和乙醇分别洗3次得到白色沉淀;60℃真空干燥12h,即得到mil-68(in)。
62.s2、合成niin ldh:取120mgs1得到的mil-68(in)分散于20ml 0.1m naoh溶液中得到混合溶液a,360mg ni(no3)2·
6h2o溶于20ml去离子水得到混合溶液b,将混合溶液a和混合溶液b充分混合,转移到50ml聚四氟乙烯内胆不锈钢反应釜中,180℃下保持反应12h;冷却至室温后,真空抽滤,并用去离子水和乙醇分别洗涤3次得到绿色沉淀;60℃真空干燥20h,得到niin ldh。
63.s3、合成niin ldh/in2s3:取60mg得到的niin ldh分散于40ml乙醇,在剧烈搅拌下加入60mg硫脲,继续剧烈搅拌30min后,转移至50ml聚四氟乙烯内胆不锈钢反应釜,100℃下保持反应2h,冷却至室温后,真空抽滤,并用去离子水和乙醇分别洗涤3次得到灰色沉淀;60℃真空干燥20h,得到niin ldh/in2s3催化剂。
64.实施例5
65.一种s型异质结催化剂的制备方法,包括如下步骤:
66.s1、合成mil-68(in):取60mg in(no3)3·
xh2o和60mg对苯二甲酸溶于40ml n,n-二甲基甲酰胺溶液,剧烈搅拌10min;然后,将溶液置于120℃油浴锅中保持30min,通过真空抽滤,并用去离子水和乙醇分别洗3次得到白色沉淀;60℃真空干燥24h,即得到mil-68(in)。
67.s2、合成niin ldh:取120mgs1得到的mil-68(in)分散于20ml 0.1m naoh溶液中得到混合溶液a,360mg ni(no3)2·
6h2o溶于20ml去离子水得到混合溶液b,将混合溶液a和混合溶液b充分混合,转移到50ml聚四氟乙烯内胆不锈钢反应釜中,180℃下保持反应12h;冷却至室温后,真空抽滤,并用去离子水和乙醇分别洗涤3次得到绿色沉淀;60℃真空干燥18h,得到niin ldh。
68.s3、合成niin ldh/in2s3:取60mg得到的niin ldh分散于40ml乙醇,在剧烈搅拌下加入60mg硫脲,继续剧烈搅拌30min后,转移至50ml聚四氟乙烯内胆不锈钢反应釜,100℃下保持反应2h,冷却至室温后,真空抽滤,并用去离子水和乙醇分别洗涤3次得到灰色沉淀;60℃真空干燥20h,得到niin ldh/in2s3催化剂。
69.对比例1
70.合成in2s3催化剂
71.取60mg in2s3分散于40ml乙醇,在剧烈搅拌下加入60mg硫脲,继续剧烈搅拌30min后,转移至50ml聚四氟乙烯内胆不锈钢反应釜,100℃下保持反应2h,冷却至室温后,真空抽滤,并用乙醇洗涤三次得到灰色沉淀;60℃真空干燥24小时,得到in2s3。
72.图2为实施例1-3制备的催化剂和对比例1的催化剂的扫描显微镜图,其中a为mil-68(in)、b为niin ldh、c为niin ldh/in2s3,(d)为对比例1制备的in2s3,从图2中可以看出,原始的mil-68(in)为形状均匀、规整的六棱柱,转化为niin ldh后转变成了2d niin ldh片层交联形成的柱状结构,硫化后的形貌良好保持了2d片层交联的柱状结构。
73.图3为实施例1-3制备的催化剂和对比例1的催化剂的x射线衍射图,其中(a)为mil-68(in)、(b)为niin ldh、niin ldh/in2s3和对比例1制备的in2s3,从图3可以看出,niin ldh/in2s3同时具有niin ldh和in2s3的x射线衍射特征峰,表明异质结的成功合成。
74.图4为实施例2-3制备的催化剂和对比例1的催化剂的拉曼光谱图,从图4可以看出,niin ldh/in2s3同时具有niin ldh和in2s3的拉曼光谱特征峰,表明异质结的成功合成。
75.图5为实施例1制备的催化剂和对比例1的x射线光电子能谱谱图,其中(a)、(b)为实施例2-3制备的niin ldh和niin ldh/in2s3和对比例1制备的in2s3催化剂光照前的xps图,(c)、(d)为实施例2-3制备的niin ldh和niin ldh/in2s3和对比例1制备的in2s3的催化剂光照下的xps图,从图5可以看出,在黑暗条件下,niin ldh/in2s3的ni 2p结合能高于niin ldh,in 3d结合能低于in2s3。光照下,niin ldh/in2s3的ni 2p结合能比未光照时更高,in2s3的in 3d结合能比未光照时更低。说明当niin ldh与in2s3接触时,电子由niin ldh转移向in2s3。而光照下,电子由in2s3转移向niin ldh。
76.图6、图7和图8分别为实施例2、实施例3和对比例1制备的催化剂的氮气吸-脱附等温线,从图6-8可以看出,异质结的比表面积明显增大,有利于催化活性位点的暴露。
77.图9为实施例2-3制备的催化剂和对比例1的催化剂的co2吸-脱附等温线,从图9可以看出,形成异质结后,co2吸附量得到明显提升。
78.图10为实施例2-3制备的催化剂和对比例1的催化剂的紫外-可见-近红外漫反射光谱,从图10可以看出,异质结结合了niin ldh和in2s3对光的捕集能力,对可见光的捕集能
力明显提升。
79.图11为实施例2制备的催化剂和对比例1的催化剂的莫特肖特曲线。从图11可以看出,niin ldh的斜率为负,表明其为n型半导体;in2s3的斜率为正,显示其为p型半导体。
80.图12为实施例2-3制备的催化剂和对比例1的催化剂的光致发光光谱和时间分辨性光致发光光谱,从图12可以看出,因为电子-空穴的结合会使能量以荧光、热量的形式耗散,较低的niin ldh/in2s3光致发光峰强度表明,与单组分催化剂相比,电子-空穴复合受到明显抑制。时间分辨性光致发光光谱得到的结论也与光致发光光谱一致,较长的衰减时间说明异质结电子-空穴复合受到明显抑制。
81.图13为实施例2-3制备的催化剂和对比例1的催化剂的瞬态光电流响应谱,从图13可以看出,较大的光电流响应说明异质结具有更好的传输动力学,有助于电子-空穴对的分离和传输。
82.图14为本发明中实施例3制备的催化剂的co与ch4产量随时间变化关系;图15为本发明中实施例2-3和对比例1的制备的催化剂的co产量对比图;图16为本发明中实施例2-3和对比例1的制备的催化剂及实施例2和对比例1催化剂共混co与ch4产量随时间变化关系;图17为本发明中实施例3制备的催化剂与其它催化剂的co产率对比图;图18为本发明中实施例3制备的催化剂在不同条件下的活性对比图;图19为本发明中实施例3制备的催化剂的co2还原循环试验;对比图14-19可以看出,niin ldh/in2s3形成异质结后,co2还原为co的活性以及选择性都得到了极大的提升,与其它s型异质结相比产率也有明显提高,并且三次循环试验证明异质结具有较好的稳定性。
83.图20为实施例2和对比例1制备的催化剂的功函数、费米能级计算结果,其中niin ldh的功函数更小、费米能级更高,当niin ldh与in2s3接触时,电子会从niin ldh转移至in2s3,与图5的xps结果相吻合。
84.图21为实施例3制备的niin ldh/in2s3的态密度计算结果,该结果说明异质结的导带活性位点为ni,在niin ldh上发生还原半反应;价带的活性位点为in,在/in2s3上发生氧化半反应。
85.图22为实施例3制备的niin ldh/in2s3的原位raman图,该结果得出了co2催化还原过程经过了*cooh、co*中间体。
86.图23为实施例3制备的niin ldh/in2s3催化剂能带结构与电子转移模型、机理示意图,当niin ldh和in2s3发生接触时,由于功函数和费米能级的不同,电子从niin ldh流向in2s3以达到相同的费能级,并形成了由niin ldh指向in2s3的内部电场。在光激发下,电子在内部电场的驱动下由in2s3转移向niin ldh,这样,使co2还原半反应发生在niin ldh上,实现了电子空穴的高效分离,提高了co2还原效率。
87.图24为实施例2-3制备的催化剂和对比例1制备的催化剂的光催化co2还原步骤自由能计算结果,与实验结果良好吻合。
88.需要说明的是,本发明中涉及数值范围时,应理解为每个数值范围的两个端点以及两个端点之间任何一个数值均可选用,由于采用的步骤方法与实施例相同,为了防止赘述,本发明描述了优选的实施例。尽管已描述了本发明的优选实施例,但本领域内的技术人员一旦得知了基本创造性概念,则可对这些实施例做出另外的变更和修改。所以,所附权利要求意欲解释为包括优选实施例以及落入本发明范围的所有变更和修改。
89.显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1