砹提纯方法
砹提纯方法
1.相关申请的交叉引用
2.根据35u.s.c.
§
119(e),本技术要求2020年4月1日提交的美国临时申请的序列号为63/003,335的优先权,其全部公开内容通过引用并入本文。
3.政府权利
4.本发明是受美国能源部科学办公室de-sc0020958下的政府支持完成的。美国政府对本发明有一定的权利。
背景技术:
5.在基于α-发射
223
racl2的成功治疗转移性去势抵抗性前列腺癌(metastatic castration-resistant prostate cancer)后,靶向α治疗(tat)药物获得了广泛的关注。有前景的性能表明需要扩大可供使用的α发射放射性核素的目录范围。一种引起广泛关注的这样的同位素是
211
at,其具有非常适合临床环境的衰变特性,其具有中等偏短的半衰期(7.2h),以及从简单的衰变纲图中获得定量的α-发射。在世界范围内只有约30台回旋加速器有能力产生可用的数量,且其中只有7台在美国,这其中只有一台目前是美国能源部同位素计划的供应商,
211
at的供应仍然有限。用能量范围为28.5-31mev的α粒子束轰击天然bi靶作为标准用于通过
209
bi(α,2n)
211
at核反应产生可用量的
211
at。尽管
211
at的有效性低,但它已经被用于许多研究治疗恶性脑肿瘤、卵巢癌的临床试验以及目前治疗晚期造血系统恶性肿瘤的研究中。
6.此外,at化学元素总体上是元素周期表上少数几个相对未被探索的领域之一。这可以归因于这样的事实:at在地球上的丰度估计仅0.07克,是所有天然存在的元素中最低的,因为at没有稳定的同位素。最长半衰期属于
210
at,仅为~8.1h,稍长于
211
at。砹是卤素系列的第五个成员,也是半金属中确认的最重的成员,这允许丰富且多样的化学。例如,观察到各种氧化态,at
–
、at0、at
+
、at
3+
、at
5+
以及at
7+
,但它们的化学和形态的详细描述受围绕其有限供应的屏障所抑制。由于其大(原子半径)、重的元素(z=85)的相对论效应而变得复杂的电子结构经历了显著的自旋-轨道-耦合,使得基于忽略自旋-轨道-耦合的计算模型对其化学行为进行预测成为问题。相反,包括自旋-轨道-耦合需要明显更多的计算资源和对模型的专门处理,以确保预测的准确性。在这样的预测中包含自旋-轨道-耦合影响at及其配合物的许多性质,包括极化率、电负性、振动频率的偏移和偶极矩的变化等等。
7.无论出于对元素周期表前沿的探索、放射性药物的应用,还是出于对at本身的理解,快速且有效的分离提纯以回收和分离这种令人感兴趣的元素都是至关重要的。历史上,采用了两种方法从bi靶中回收
211
at:干法蒸馏和湿化学处理。后者已被证明能产生更具重现性的
211
at回收率。对于分析尺度分离,其中待回收和提纯的
211
at的量为约1-10ng,大量的bi(1-10g)构成基体的大部分,溶剂萃取本身不能作为有效的分离方法,因为它仅限于每次接触单个分离阶段,并且需要复杂的设备来以连续流动的方式运行。相反,色谱可以在单个柱中提供大量的阶段,并且本质上以连续流动的方式运行。
8.both woen等人(inorg.chem.59(2020)6137
–
6146)以及li等人(sci.rep.9(2019)16960)最近展示了从轰击的
209
bi靶中回收
211
at的有效色谱系统,使用预过滤树脂和使用碲金属粉末的产率分别为68%和95%。us 2018/0308599还描述了使用色谱分离
211
at的方法。然而,这些方法要求将体系从硝酸盐转化为氯化物介质,这增加了一个缓慢、耗时的步骤,即要么蒸发至干燥以去除硝酸盐,要么该方法需要用氯化羟胺化学破坏硝酸盐。因此,需要一种快速且更有效的方法来回收at。
技术实现要素:
9.根据本公开的一方面,一种方法包括:
10.(a)使包含砹和铋的组合物与硝酸接触以形成包含砹、铋和硝酸的第一溶液;
11.(b)使树脂与所述第一溶液接触,从而使砹从所述第一溶液中分离出来并进入所述树脂中;以及
12.(c)从树脂中洗脱砹。
13.在另一方面,组合物包含ato
+
x-,其中x是反离子。
14.从以下的详细描述并通过本公开的实践,本公开的另外的实施方案、特征和优势将是显而易见的。本公开的方法和化合物可以描述为以下列举条款中任一条款的实施方案。应当理解,在这些实施方案彼此不矛盾的情况下,本文描述的实施方案中的任一个可以与本文描述的任何其他实施方案结合使用。
15.1.一种方法,其包括
16.(a)使包含砹和铋的组合物与硝酸接触以形成包含砹、铋和硝酸的第一溶液;
17.(b)使树脂与所述第一溶液接触,从而使砹从所述第一溶液中分离出来并进入所述树脂中;以及
18.(c)从所述树脂中洗脱砹。
19.2.根据条款1所述的方法,其中用溶剂浸渍树脂。
20.3.根据条款1或2所述的方法,其中所述溶剂包含有机溶剂。
21.4.根据前述条款中任一项所述的方法,其中所述有机溶剂是极性的。
22.5.根据前述条款中任一项所述的方法,其中所述有机溶剂包含任选取代的c
1-c
18
烷基。
23.6.根据前述条款中任一项所述的方法,其中所述有机溶剂包含羰基。
24.7.根据前述条款中任一项所述的方法,其中所述有机溶剂包含醛、酮、酯、酰胺、碳酸酯、羧酸酯或氨基甲酸酯。
25.8.根据前述条款中任一项所述的方法,其中所述有机溶剂为化学式c
1-c6烷基-c(o)-c
1-c6烷基,其中c
1-c6烷基中的每个氢原子任选地被取代。
26.9.根据前述条款中任一项所述的方法,其中所述有机溶剂是辛酮。
27.10.根据前述条款中任一项所述的方法,其中所述有机溶剂是3-辛酮。
28.11.根据条款1-5中任一项所述的方法,其中所述有机溶剂是c
1-c
18
烷醇。
29.12.根据前述条款中任一项所述的方法,其中所述砹在所述有机溶剂中的d值分配系数为至少20。
30.13.根据前述条款中任一项所述的方法,其中所述砹在所述有机溶剂中的d值分配
系数为至少40。
31.14.根据前述条款中任一项所述的方法,其中所述砹在所述有机溶剂中的d值分配系数为至少60。
32.15.根据前述条款中任一项所述的方法,其中所述砹在所述有机溶剂中的d值分配系数为至少80。
33.16.根据前述条款中任一项所述的方法,其中所述树脂是惰性树脂。
34.17.根据前述条款中任一项所述的方法,其中所述树脂是聚合物树脂、沸石、分子筛或多孔玻璃珠。
35.18.根据前述条款中任一项所述的方法,其中所述树脂包括苯乙烯-二乙烯基苯共聚物。
36.19.根据条款18所述的方法,其中所述苯不含官能团。
37.20.根据前述条款中任一项所述的方法,其中所述砹为
211
at。
38.21.根据前述条款中任一项所述的方法,其中所述砹为
209
at。
39.22.根据前述条款中任一项所述的方法,其中所述铋不分离到树脂中。
40.23.根据前述条款中任一项所述的方法,其中所述第一溶液中的硝酸的浓度为约1m至约10m。
41.24.根据前述条款中任一项所述的方法,其中所述第一溶液中的硝酸的浓度为约1m至约8m。
42.25.根据前述条款中任一项所述的方法,其中所述第一溶液中的硝酸的浓度为约2m至约8m。
43.26.根据前述条款中任一项所述的方法,其中所述方法进一步包括在步骤(b)之后洗涤所述树脂的步骤。
44.27.根据前述条款中任一项所述的方法,其中洗涤步骤通过使水溶液通过所述树脂来进行。
45.28.根据条款27所述的方法,其中所述水溶液包含酸。
46.29.根据条款28所述的方法,其中所述酸是硝酸、氢溴酸、盐酸、硫酸或高氯酸。
47.30.根据条款28或29所述的方法,其中所述酸的浓度为约1m至约10m。
48.31.根据前述条款中任一项所述的方法,其中所述方法从混合物中回收≥85%的砹。
49.32.根据前述条款中任一项所述的方法,其中所述方法从中回收≥90%的砹。
50.33.根据前述条款中任一项所述的方法,其中所述方法从混合物中回收≥95%的砹。
51.34.根据前述条款中任一项所述的方法,其中步骤(c)后的砹的纯度≥90%。
52.35.根据前述条款中任一项所述的方法,其中步骤(c)后的砹的纯度≥95%。
53.36.根据前述条款中任一项所述的方法,其中步骤(c)后的砹的纯度≥99%。
54.37.根据前述条款中任一项所述的方法,其中洗脱步骤通过使树脂与第二有机溶剂接触来进行。
55.38.根据条款37所述的方法,其中第二有机溶剂包含丙酮或c
1-c
18
烷醇。
56.39.根据条款38所述的方法,其中第二有机溶剂包含乙醇。
57.40.根据条款37-39中任一项所述的方法,其中第二有机溶剂可与第一有机溶剂混溶。
58.41.根据前述条款中任一项所述的方法,其中步骤(a)、(b)和(c)在少于约1小时内进行。
59.42.根据前述条款中任一项所述的方法,其中步骤(a)、(b)和(c)在少于约30分钟内进行。
60.43.根据前述条款中任一项所述的方法,其中步骤(a)、(b)和(c)在少于约15分钟内进行。
61.44.根据前述条款中任一项所述的方法,其中步骤(a)、(b)和(c)在少于约10分钟内进行。
62.45.根据前述条款中任一项所述的方法,其中步骤(a)、(b)和(c)在少于砹半衰期的约20%内进行。
63.46.根据前述条款中任一项所述的方法,其中步骤(a)、(b)和(c)在少于砹半衰期的约15%内进行。
64.47.根据前述条款中任一项所述的方法,其中步骤(a)、(b)和(c)在少于砹半衰期的约10%内进行。
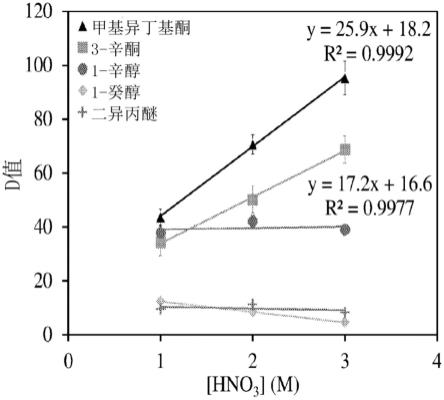
65.48.根据前述条款中任一项所述的方法,其中步骤(a)、(b)和(c)在少于砹半衰期的约5%内进行。
66.49.根据前述条款中任一项所述的方法,其中所述方法包括在步骤(b)之前制备所述树脂的步骤。
67.50.根据条款49所述的方法,其中制备所述树脂的步骤包括使所述树脂与有机溶剂接触。
68.51.根据前述条款中任一项所述的方法,其中所述方法进一步包括用洗脱的砹标记治疗剂的步骤。
69.52.一种包含ato
+
x-的组合物,其中x-是反离子。
70.53.根据条款52所述的组合物,其中x-是硝酸根、卤离子或高氯酸根。
71.54.根据条款53所述的组合物,其中x-是硝酸根。
72.55.根据条款53所述的组合物,其中x-是高氯酸根。
73.56.根据条款53所述的组合物,其中x-是卤离子。
74.57.根据条款56所述的组合物,其中所述卤离子是氯离子。
75.58.根据条款52-57中任一项所述的组合物,其中所述组合物与有机溶剂络合。
76.59.根据条款58所述的组合物,其中所述有机溶剂包含任选取代的c
1-c
18
烷基。
77.60.根据条款58所述的组合物,其中所述有机溶剂包含羰基。
78.61.根据条款58所述的组合物,其中所述有机溶剂包含醛、酮、酯、酰胺、碳酸酯、羧酸酯或氨基甲酸酯。
79.62.根据条款58所述的组合物,其中所述有机溶剂为化学式c
1-c6烷基-c(o)-c
1-c6烷基,其中c
1-c6烷基中的每个氢原子任选地被取代。
80.63.根据条款58所述的组合物,其中所述有机溶剂是辛酮。
81.64.根据条款58所述的组合物,其中所述有机溶剂是3-辛酮。
82.65.根据条款52-64中任一项所述的组合物,其中所述砹为
211
at。
83.66.根据条款52-64中任一项所述的组合物,其中所述砹为
209
at。
84.67.根据条款52-66中任一项所述的组合物,其由根据条款1-51中任一项所述的方法生产。
85.68.一种组合物,其包括通过根据条款1-51中任一项所述的方法生产的砹。
86.69.一种方法,其由条款1-51中任一项所述的步骤组成。
87.70.一种方法,其基本上由条款1-51中任一项所述的步骤组成。
88.在考虑到举例说明如当前所理解的实施本公开的最佳方式的说明性实施方案时,本公开的另外的特征对于本领域技术人员将变得显而易见。
附图说明
89.图1示出了
211
at萃取到不同有机溶剂中的d值作为初始hno3水溶液浓度的函数。实线用于视觉辅助。注:在所有情况下,bi的d值≤0.05。
90.图2示出了树脂在用1-辛醇和3-辛酮浸渍之前(蓝色)以及浸渍之后的tga曲线。
91.图3示出了根据toc分析计算出的从3-辛酮浸渍的树脂床(0.5-ml bv,7mm id
×
13mm高)浸出到收集部分中的3-辛酮浸出的量(
■
)和总浸出的百分比(
◆
)。箭头表示每个数据集的对应轴。
92.图4示出了用1-辛醇浸渍的树脂床(0.5-ml bv,7mm id
×
13mm高)的2m hno3的0.5-ml等分试样(aliquots)的色谱图,该等分试样含有20-μl第1回溶解的轰击靶溶液的掺料(spike)(约13μci
211
at)。注:数据进行了衰变校正,以说明半衰期的差异;bi由icp-ms测定。
93.图5示出了用3-辛酮浸渍的树脂床(0.5-ml bv,7mm id
×
13mm高)的2m hno3的0.5-ml等分试样的色谱图,该等分试样含有20-μl第1回溶解的轰击靶溶液的掺料(约13μci
211
at)。注:数据进行了衰变校正,以说明半衰期的差异;bi由icp-ms测定。
94.图6示出了用3-辛酮浸渍的树脂床(0.5-ml bv,7mm id
×
13mm高)的6m hno3的0.5-ml等分试样的色谱图,该等分试样含有20-μl第1回溶解的轰击靶溶液的掺料(约13μci
211
at)。注:数据进行了衰变校正,以说明半衰期的差异;bi由icp-ms测定。
95.图7示出了用3-辛酮浸渍的树脂床(0.5-ml bv,7mm id
×
13mm高)的2m hno3的1.5-ml等分试样的色谱图,该等分试样含有399-μl第2回溶解的轰击靶的掺料(约1.0mci
211
at)以及42-μl 207
bi的掺料(约10nci)。注:死体积被假定为bv的一半,但似乎已经过高估计,因为在部分中观察到少量的
207
bi和
66/67
ga;数据经衰变校正,以说明半衰期的差异。
96.图8示出了用3-辛酮浸渍的树脂床(0.5-ml bv,7mm id
×
13mm高)的4m hno3的1.4-ml等分试样的色谱图,该等分试样含有399-μl第2回溶解的轰击靶的掺料(约1.0mci
211
at)以及42-μl 207
bi溶液的掺料(约10nci)。注:死体积被假定为bv
的一半,但似乎已经过高估计,因为在部分中观察到少量的
207
bi和
66/67
ga;数据经衰变校正,以说明半衰期的差异。
97.图9示出了用3-辛酮浸渍的树脂床(0.5-ml bv,7mm id
×
13mm高)的5.7m hno3的1.3-ml等分试样的色谱图,该等分试样含有399-μl第2回溶解的轰击靶的掺料(约1.0mci
211
at)以及42-μl 207
bi的掺料(约10nci)。注:死体积被假定为bv的一半,但似乎已经过高估计,因为在部分中观察到少量的
207
bi和
66/67
ga;数据经衰变校正,以说明半衰期的差异。
98.图10示出了用3-辛酮浸渍的树脂床(0.5ml bv,7mm id
×
13mm高)的第1回溶解的轰击靶溶液(4.1mci 211
at在约6m hno3中)的5-ml等分试样的色谱图。注:数据经衰变校正,以说明半衰期的差异;采用icp-ms法测定bi。
99.图11示出了用3-辛酮浸渍的树脂床(0.5ml bv,7mm id
×
13mm高)的5.9m hno3的4-ml等分试样的色谱图,该等分试样含有3.76-ml第2回溶解的轰击靶溶液的掺料(约9.8mci 211
at)和240μl 207
bi的掺料(约57.6nci)。注:死体积被假定为bv的一半,但似乎已经过高估计,因为在部分中观察到少量的
207
bi和
66/67
ga;数据经衰变校正,以说明半衰期的差异。
具体实施方式
100.砹(at)可用作放射性标记用于治疗。然而,at的天然丰度较低。at可以通过用α粒子轰击铋(bi)金属靶而产生。产生的at随后必须与未反应的bi分离。本文描述了一种使用色谱法从组合物(例如由轰击bi形成的组合物)中分离at的方法。在说明性实施方案中,本文描述的方法溶解含有at的组合物,并随后从溶解的混合物中分离at。所描述的方法可以在不需要转换介质或用于最初溶解at/bi组合物的溶液的情况下进行。
101.本文描述的at可以是
209
at或者
211
at及其阳离子物种。例如,本文方法中描述的at可以是阳离子物种ato
+
,但仍被称为at。说明性地,该方法可包括产生at。at可以由
209
bi(α,2n)
211
at核反应(其中用α粒子轰击
209
bi金属)产生。所形成的轰击的靶可以包括at、未反应的bi以及副产物的混合物。
102.在一些方面,从包含bi和at的组合物中分离at。说明性地,使组合物与溶液(如水溶液)接触。在说明性方面,该水溶液包括酸,例如有机酸或无机酸。无机酸可以是硝酸。该溶液溶解或基本上溶解该组合物以形成包含at和bi的溶液。在一些实施方案中,该溶液包含at、bi和酸。在一些实施方案中,该溶液包含at、bi和硝酸。
103.在一些方面,该溶液具有特定浓度的酸,或者在后续步骤之前调节该溶液以具有特定浓度的酸。说明性地,酸可以帮助溶解组合物。例如,硝酸的存在可以有助于溶解轰击的bi靶。
104.说明性地,酸的浓度可以是约1m至约10m、约1m至约8m、约2m至约8m或约3m至约7m。酸的浓度可以是约1m、约2m、约3m、约4m、约5m、约6m、约7m、约8m、约9m或约10m。酸的浓度可以根据在后续步骤中使用的溶剂中的分配系数来调节。当酸是有机酸或无机酸(如硝酸)时,本文描述的范围同样适用。
105.在一些方面,at用色谱法分离。在说明性实施方案中,通过使用树脂进行色谱法。树脂可以是树脂床的形式。树脂床可以在柱中。可选地,树脂可用于批量处理。说明性树脂
包括聚合物树脂和玻璃树脂。在一些实施方案中,树脂包括沸石、分子筛、聚合物树脂或玻璃树脂。在一些实施方案中,树脂是多孔的。在一些实施方案中,多孔树脂是聚丙烯酸酯树脂或多孔玻璃珠。树脂可以是惰性的。在一些实施方案中,树脂包括苯乙烯-二乙烯基苯共聚物。在一些实施方案中,共聚物的苯不含官能团。
106.在一些方面,该方法包括制备树脂的步骤。该步骤可以在树脂与包含at的溶液接触之前进行。在说明性实施方案中,树脂可以与溶剂接触,例如有机溶剂。说明性地,制备树脂的步骤得到用溶剂(例如有机溶剂)浸渍的树脂。在一些实施方案中,有机溶剂是极性的。在一些实施方案中,有机溶剂包括任选取代的c
1-c
18
烷基,其中c
1-c
18
烷基的每个氢原子任选地被官能团取代。c
1-c
18
烷基上的任选取代基是本领域公知的,且包括卤素、羟基、胺基、硫醇基、氧代基(oxo)、酮基、羧酸酯基、醛基、酰胺基、碳酸酯基、氨基甲酸酯基及其组合等。在一些实施方案中,有机溶剂包括醛、酮、酯、酰胺、碳酸酯、羧酸酯或氨基甲酸酯。在一些实施方案中,有机溶剂包含,包括醛基、酮基、酯基、酰胺基、碳酸酯基、羧酸酯基或氨基甲酸酯基的c
1-c
18
,c
1-c
12
或c
1-c6烷基。在一些实施方案中,有机溶剂为化学式c
1-c6烷基-c(o)-c
1-c6烷基。在一些实施方案中,有机溶剂为化学式c
1-c6烷基-c(o)-c
1-c6烷基,其中c
1-c6烷基中的每个氢原子任选地被取代。在一些实施方案中,有机溶剂是辛酮。在一些实施方案中,有机溶剂是3-辛酮。在一些实施方案中,有机溶剂是c
1-c
18
烷醇。说明性地,有机溶剂可以包括本文所述有机溶剂的混合物。
107.术语“烷基”是指直链或支链的单价烃基。在一些实施方案中,有利的是将“烷基”中的原子数限制在原子的特定范围内,例如c
1-c
18
烷基、c
1-c
12
烷基或c
1-c6烷基。烷基的实例包括甲基(me)、乙基(et)、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基(tbu)、戊基、异戊基、叔戊基、己基、异己基,以及根据本领域普通技术和本文提供的教导可认为与前述实例中的任何一个基团等效的基团。应当理解,烷基可以是未取代的或如本文所述的取代的。烷基可以被本文所述的各种实施方案中的任何取代基取代,包括一个或多个这样的取代基。术语“烷
‑”
可以形成前缀,其余部分是官能团。例如,“烷醇”是被醇取代的烷基。
108.术语“取代的”是指特定的基团或部分带有一个或多个取代基。术语“未取代的”是指特定的基团没有取代基。当术语“取代的”用于描述一个结构体系时,该取代意味着发生在该体系上任何化合价允许的位置。在一些实施方案中,“取代的”意味着特定基团或部分具有一个、两个或三个取代基。例如,烷基的碳上的两个氢原子可以被氧代(=o)基取代以形成羰基(c=o)。在其他实施方案中,“取代的”是指特定基团或部分带有一个或两个取代基。在其他实施方案中,“取代的”是指特定基团或部分带有一个取代基。
109.术语“卤素”或“卤代(halo)”指氯、氟、溴或碘。
110.在一些方面,浸渍树脂的溶剂是提供at相对于水溶液(例如包含硝酸的水溶液)的d值分配系数至少为10的溶剂。在说明性实施方案中,at在有机溶剂中的d值分配系数为至少约20、至少约40、至少约60、或至少约80。说明性地,分配系数可以相对于包含酸(例如硝酸)的水溶液来测量。在一些实施方案中,at在辛酮和包含约2-6m硝酸的水溶液之间的分配系数为至少约20或至少约40。
111.在一些实施方案中,at组合物以与树脂床体积成比率的溶液体积装载到树脂上。在一些实施方案中,含at的溶液以至高约10、至高约8、至高约6或至高约4床体积的比率装载到树脂床上。用于装载at组合物的床体积的数量可以通过本领域内已知的方法来调节,
以使分离到树脂中的at的量最大化。
112.说明性地,at可以接触树脂,并且at可以分离到树脂中,形成络合物,或者与树脂形成有利的相互作用。说明性地,尽管bi可以接触树脂,但bi不会保留在树脂中或在树脂上。或者,bi可以与树脂形成弱相互作用,使得当洗涤树脂时,结合或保留的bi被除去。应当理解,接触包括任何方式的化学相互作用,例如离子或非共价相互作用。应进一步理解,分离到树脂中包括分离到树脂的内部空间中以及接触树脂的表面并形成有利的相互作用以将at保持在树脂上。
113.在与含有at的混合物的溶液接触的步骤之后,可以洗涤树脂。洗涤步骤可以包括用水溶液洗涤树脂。在一些实施方案中,水溶液包括酸。在一些实施方案中,水性洗涤溶液包含浓度低于用于溶解at/bi组合物的酸浓度的酸。例如,如果在溶解at/bi组合物的水溶液中酸浓度约为6m,洗涤溶液中的酸浓度可以小于约6m,例如约2m。在一些实施方案中,酸的浓度小于约10m,小于约8m,小于约6m,或小于约4m。在一些实施方案中,酸的浓度至高约8m,至高约6m,或至高约4m。在说明性实施方案中,洗涤步骤中使用的酸(例如硝酸)与前面步骤中的酸相同。
114.或者,酸可以是不同的酸。例如,酸可以是hclo4,,hcl,hbr,或h2so4。说明性地,改变洗涤步骤中使用的酸可以改变洗脱步骤回收的分离的at的反离子。
115.在一些实施方案中,洗涤树脂的步骤通过床体积比来测量。例如,树脂可以用体积为至少约2、至少约3、至少约4、至少约5或至少约6床体积的溶液洗涤。在说明性实施方案中,树脂可以用含有酸的水溶液和不含酸的水溶液依次洗涤。洗涤步骤可以通过本领域已知的方法来调节,以包括额外的或更少的水溶液洗涤步骤。
116.所述方法包括从树脂中洗脱at的步骤。说明性地,洗脱步骤使at从树脂中解离,以使at被收集。洗脱步骤可以通过将树脂与有机溶剂接触来进行。在一些实施方案中,有机溶剂是与浸渍树脂相同的溶剂。在一些实施方案中,洗脱步骤中的有机溶剂可与浸渍树脂的溶剂混溶。在一些实施方案中,有机溶剂包括任选取代的c
1-c
18
烷基,其中c
1-c
18
烷基的每个氢原子任选地被官能团取代。c
1-c
18
烷基上的任选的取代基是本领域公知的,且包括卤素、羟基、胺基、硫醇基、氧代基、酮基、羧酸酯基、醛基、酰胺基、碳酸酯基、氨基甲酸酯基及其组合等。在一些实施方案中,有机溶剂包括醛、酮、酯、酰胺、碳酸酯、羧酸酯或氨基甲酸酯。在一些实施方案中,有机溶剂包含包括醛基、酮基、酯基、酰胺基、碳酸酯基、羧酸酯基或氨基甲酸酯基的c
1-c
18
,c
1-c
12
或c
1-c6烷基。在一些实施方案中,有机溶剂为化学式c
1-c6烷基-c(o)-c
1-c6烷基。在一些实施方案中,有机溶剂为化学式c
1-c6烷基-c(o)-c
1-c6烷基,其中c
1-c6烷基中的每个氢原子任选地被取代。在一些实施方案中,有机溶剂是辛酮。在一些实施方案中,有机溶剂是3-辛酮。在一些实施方案中,有机溶剂是c
1-c
18
烷醇。在一些实施方案中,溶剂包括乙醇。
117.说明性地,本文所述方法从包含at的组合物中回收至少约80%、至少约85%、至少约90%或至少约95%的at。在一些实施方案中,该方法从包含at的组合物中回收约80%至约99%、约85%至约99%或约90%至约99%的at。
118.说明性地,洗脱的at中at的纯度高于在色谱步骤之前包含at的组合物。在一些实施方案中,洗脱的at的纯度为至少约90%、至少约95%或至少约99%。
119.本文所述的方法可在少于约1小时、少于约30分钟、少于约15分钟或少于约10分钟
内进行。说明性地,与需要在色谱法之前将at/bi组合物溶解在硝酸溶液中、蒸发硝酸溶液并在盐酸中重构残留物的步骤的比较方法相比,或者与需要在色谱法之前破坏t硝酸的比较方法相比,该方法在更短的时间内进行。本文所述的方法在其半衰期的特定百分比内分离at。在一些实施方案中,该方法在at(例如
211
at)的半衰期的小于约20%、小于约15%、小于约10%或小于约5%内进行。
120.在一些实施方案中,该方法可进一步包括用洗脱的at标记治疗剂。这可以直接用洗脱的at的柱部分进行,或者可以包括浓缩含at的部分,并将at悬浮或溶解在用于标记步骤的溶液中。
121.在另一方面,一种组合物包含盐ato
+
x-,其中x-是反离子。反离子可以是本文所述方法中使用的水性酸的共轭碱,或者可以是任何合适的酸的共轭碱。在一些实施方案中,x-是硝酸根离子、卤素离子或高氯酸根离子。在一些实施方案中,x-是硝酸根离子。在一些实施方案中,x-是卤素离子。合适的卤素离子包括氟离子、氯离子或溴离子。在一些实施方案中,x-是高氯酸根离子。在一些实施方案中,ato
+
的at是
211
at或者
209
at。
122.实施例
123.材料:
124.硝酸(67-70%hno3)购自bdh chemicals;3-辛酮(acs级≥96%)购自emd millipore corp.;1-辛醇(实验室级)购自ward's science;乙醇(≥99.5%200度(proof))购自emd,且全部按来样(as received)使用。从elga labwater purelab flex ultrapure实验室水净化系统中获得去离子(di)水,该系统在25℃下以18.2mωcm运行。
125.铋-207购自eckert&ziegler isotope products(valencia,ca),为bi(no3)3溶液,其每毫升约含0.24μci,且在4m hno3中bi总浓度为约48μm。
126.方法:
127.使用thermo fisher scientific icap rq质谱仪,采用电感耦合等离子体质谱法(icp-ms)对bi进行定量分析。使用相对效率为20.0%且工作探测器体积约为115cm3的校准的canberra model gc2020高纯度锗探测器(hpge),以及inspector
tm 2000数字信号分析仪(dsa,canberra industries inc.,meriden,ct)和genie-2000软件通过伽马(γ)射线光谱法进行
207
bi的半定量分析和
211
at的定量分析。探测器在122kev处的能量分辨率为1.0kev,在1300kev处的能量分辨率为1.98kev。相关的核数据从browne和firestone获得。所有的校准都用购自eckert&ziegler isotope products的可溯源至美国国家标准与技术研究所(nist)的
152
eu标准γ-射线源确定。用1064kev的γ-射线直接追踪
207
bi。用76.9kev、79.9kev、89.8kev、92.3kev的x-射线和687kev的γ-射线直接追踪
211
at。杂质(
66/67
ga)通过半衰期分析(见si)鉴定,并用833kev和1039kev的γ-射线直接追踪
66
ga,用185kev和300kev的γ-射线直接追踪
67
ga。
128.at-211的生产:
129.通过
209
bi(α,2n)
211
at核反应在两个单独的回合中产生砹-211,该核反应通过在texas a&m k150回旋加速器上28.8mev的α-粒子轰击(截面约为0.9靶恩(barn))[7],天然bi金属靶(同位素纯
209
bi,金属纯度≥99.997%,购自goodfellow)9-10小时,其平均束流为2.4-3.2pμa。bi金属靶的质量约为9.4g或1.0g,呈跑道椭圆形(6.985
×
1.27cm),两端各覆盖有半圆(半径0.635cm),其厚度估计为950μm或100μm,该金属钯安装在铝框架(6061铝合
金,95%铝)中,与冷却至15℃的再循环水冷却的支撑块接触。靶保持在与束成10
°
角,以使靶的覆盖最大化,同时使束至铝外壳的损失最小化。轰击的靶溶解在11.2m或8m的hno3中,使得最终的hno3浓度为约6m。然后对溶液进行取样,并在轰击结束时测定
211
at的生产产率。
211
at和
207
bi在较小程度上是高放射性的,其根据alara原则在配备适当处理放射性物质的实验室中处理,使用了放射性生物安全柜。
[0130]
分离:
[0131]
研究了从不同浓度的hno3到多种有机溶剂中的一系列萃取。首先,用简单的直链醇,1-辛醇和1-癸醇从1-3m hno3中萃取
211
at,如图1所示。在硝酸浓度为1、2和3m时,将
211
at萃取到1-辛醇中得到的分配比(d)值分别为约37.9
±
2.3、42.0
±
2.2和38.9
±
2.1。因此,最大萃取发生在约2m hno3。与此相反,
207
bi在所有三种酸度下进入1-辛醇的d值均≤0.05,这与水相中存在的所有活性相对应,而在有机相中
207
bi的量低于检测限。将脂肪链长度从c8增加到c
10
对
211
at的萃取有负面影响,且在1m hno3中的d值为12.3
±
0.8,2m hno3中的d值为8.4
±
0.4,3m hno3中的d值为4.6
±
0.2,萃取率分别降低了约3倍、5倍和9倍。此外,到1-癸醇中的最大萃取出现在≤1m hno3时,因为随着hno3浓度从1m增加到3m,d值呈线性下降。
207
bi萃取到1-癸醇中的d值≤0.05。虽然与hno3一起的金属萃取的确切机理仍是未知,但1-癸醇的更具非极性性质似乎抑制了萃取。这可能是源于维持电荷平衡的需要和将硝酸根抗衡阴离子与阳离子at物种一起共萃取到有机相中的结果。假设at(iii),即ato
+
分子阳离子是被萃取的物种,以下的平衡描述了萃取:
[0132][0133]
为了进一步验证这一点,研究了较低极性的溶剂(二异丙醚)和较高极性的溶剂(甲基异丁基酮)。
211
at到二异丙醚中的萃取在1-癸醇的范围内,
207
bi继续保留在水相中(d值≤0.05)。与研究的其它溶剂体系相比,
211
at在甲基异丁基酮体系中表现出明显的不同,表现出强的hno3依赖性。在1m hno3中,
211
at到甲基异丁基酮中的萃取率略高于1-辛醇,当hno3浓度增加到2m和3m时,d值分别增加了约1.7x和2.4x。另一方面,
207
bi表现出与所研究的其他体系相似的行为,其d值非常低(≤0.05)。对具有与1-辛醇相似极性的第二种酮(3-辛酮)进行测试,以确定更具极性的甲基异丁基酮的溶剂化作用是否是萃取的驱动力,或者酮的羰基官能团是否起主要作用。与甲基异丁基酮一样,在1m hno3中
211
at到3-辛酮中的萃取与1-辛醇的萃取类似,当hno3浓度增加到2m和3m时,d值分别增加了约1.2x和1.8x。同样,
207
bi仍保留在水相中(d值≤0.05)。用dft计算证明了酮对ato
+
的强化萃取优于醇,该计算显示丙酮的结合自由能比异丙醇强4.6kcal mol
–1。
[0134]
211
at在3-辛酮和甲基异丁基酮体系中的萃取总体行为是相似的,显示出
211
at的d值与在1-3m hno3间的水相中的初始hno3浓度呈线性关系,而甲基异丁基酮体系的斜率比3-辛酮体系陡约50%。作为hno3浓度的函数,进入甲基异丁基酮和3-辛酮的
211
at的d值之间的直接相关性可能表明酮与at金属中心之间的相互作用。目前,这样的相互作用的性质尚不完全清楚。密度泛函计算显示了ato
+
的空π*轨道与丙酮的o的
‘
sp2’
杂化孤对电子之间的强的供体-受体相互作用。对ato
+
_异丙醇的nbo分析表明,ato
+
_异丙醇的o的sp3杂化孤对电子向ato
+
提供的电子比ato
+
_丙酮的o的sp2杂化孤对电子轨道提供的少0.11。这种相互作用比ato
+
与异丙醇的o的
‘
sp3’
杂化孤对电子的相互作用强4.6kcal/mol,而ato
+
_丙酮的溶剂校
正的gibbs结合自由能比ato
+
_异丙醇高2.1kcal/mol。因此,酮表现出与ato
+
的强的结合,这导致更好的萃取。在h2o-有机界面上,有机分子的极性端(氧)在h2o层中。ato
+
和no3–
在h2o层中会被溶剂分离,所以这些物种在萃取ato
+
时的早期相互作用将是ato
+
与有机分子中的氧结合。ato
+
进入有机层的运动必然需要伴随no3–
。
[0135]
萃取色谱法:
[0136]
cg300m多孔珠(孔体积为0.7ml g-1
,粒径为50-100μm),在20%乙醇中形成浆料,购自sigma-aldrich。cg300m是苯乙烯-二乙烯基苯共聚物,其苯环上没有官能团。在使用之前,将珠在80℃下干燥至少24h,以去除孔隙中的任何溶剂。然后通过将珠浸泡在有机溶剂中不少于24小时,用1-辛醇或3-辛酮浸渍干燥的树脂。用ta仪器tga 5500在n2流下以10℃min-1
的加热速率对浸渍的树脂和干燥的树脂进行热重分析(tga)。然后将浸渍的树脂装入2-ml的中,其床体积(bv)为0.5ml,内径(id)为0.7cm。过量的有机溶剂保持在柱中在浸渍的树脂床上方,以防止溶剂从孔中蒸发。在萃取色谱分离之前,从柱中排出过量的溶剂。
[0137]
一般的色谱法程序如下。将装载溶液(0.5-5ml在2-6m的hno3中掺有13
±
1.3μci至9.8
±
0.98mci的
211
at的溶液)以0.5ml等分试样通过柱,接下来4个2m hno3的0.5-ml等分试样通过柱,随后是h2o的0.5ml等分试样,最后是3-5个乙醇的0.5ml等分试样。用注射器对柱的顶部空间手动加压,以加快每个部分的洗脱。在所有情况下,每0.5ml收集一次部分,并用γ-射线光谱学分析每个部分。采用shimadzutoc-vwp分析仪对从用3-辛酮浸渍的树脂(bv约0.5ml)填充的色谱柱中洗脱的部分进行总有机碳分析(toc);然而,没有放射性物种存在。
[0138]
柱表征:
[0139]
如图2所示,用tga测定用1-辛醇或3-辛酮浸渍的树脂的装载量。干燥的未浸渍的树脂在加热到200℃时重量损失很小,为2.44%,这表明只有从大气中吸收的表面h2o。来自水的低质量百分比并不令人惊讶,因为本质上是疏水性的,其具有悬挂在孔隙空间中的苯基官能团。另一方面,浸渍的树脂在200℃下的重量损失要大得多,其中对于1-辛醇和3-辛酮的重量损失分别为70.2%和65.5%。这表明孔隙已经被有机溶剂填充。然而,由于相对较低的沸点和较高的蒸气压(见表1),除去溶剂所需的低温使得珠必须保持浸没在溶剂中直至即将使用之前。
[0140]
表1:所选有机溶剂的性质。
[0141][0142][0143]
为了确定有机溶剂在色谱法过程中是否从孔隙中浸出,对6m hno3、2m hno3和h2o洗脱液的部分进行了总有机碳(toc)分析,这些洗脱液依次通过用3-辛酮浸渍的珠填充的萃取色谱法系统,以得到0.5ml的bv。图3示出了每个部分中的3-辛酮的量和基于珠密度的浸出百分比。3-辛酮浸渍的珠密度计算为0.90
±
0.05g/ml,如下所示。用
0.45μm醋酸纤维素离心管过滤器和微型离心机离心,将浸渍的珠与过量的3-辛酮溶剂分离,并称重。然后将已知体积的3-辛酮加入到珠中,并测量体积位移,如下等式所示。
[0144][0145]
在前两个部分中观察到少量的3-辛酮,分别为总量的1.4%和0.9%,而随后的部分处于或低于基线。6m hno3的前两个部分中3-辛酮的初始浸出可能是由于粒间间隙空间中残留的溶剂,而不是孔隙中吸附的溶剂。这表明在萃取色谱法过程中,有机溶剂的很少的浸出是从树脂的孔隙中除去的。在有意洗脱核素之前,
211
at的渗出很少,这主要表明
211
at被萃取到浸渍的溶剂中,并且含有萃取的
211
at的溶剂保留在孔隙中。基于二氧化硅支架的其它萃取色谱法体系显示出更高的关于有机相浸出倾向,并且需要加入多组分溶剂体系来改善固定相的疏水性特性。
[0146]
萃取色谱法:
[0147]
为了使
211
at在制药装置中使用,其需要从非放射性的天然bi和其生产过程中产生的任何放射化学污染物中提纯。如前所述,
211
at的半衰期短(t
1/2
约7.2h)需要快速化学来实现与主基体(本例中是bi金属)的分离。首先,用少量掺料的在2m hno3中的第1回溶解的轰击靶溶液(约13μci,
211
at)对bv为0.5ml的1-辛醇萃取色谱系统进行了测试。如图4中的色谱图所示,
211
at几乎被定量萃取(≥97%)到柱上,而大约73%的bi(在1-辛醇中的d值≤0.1)和大部分的放射化学污染物没有被保留,通过柱并在装载部分中洗脱。bi和污染物物种一起在2m hno3洗涤中被进一步洗脱,回收洗涤的第一部分中的剩余部分。应注意,没有尝试调整部分收集以对应于装载溶液前端。也就是说,一旦装载溶液被添加到柱的顶部,洗脱的每一滴都被收集。因此,溶液前200-250μl对应于柱的自由体积(bv的40-50%)中剩余溶液的前200-250μl。这表明,虽然在收集的第一洗涤液部分中看到了bi和放射化学污染物,但它们很可能完全不受抑制地通过柱,仅留在初始装载溶液中。随后的2m hno3洗涤液的三个部分中没有任何这些物种。另一方面,
211
at大部分保留在柱上,整个洗涤过程中仅大约5%被洗脱。与之后的洗涤液部分一样,h2o冲洗液仅含有微量的
211
at,且不含任何其他令人感兴趣的物种。然后尝试用乙醇剥离柱,在这样做时,溶解含有萃取的
211
at的有机溶剂,这将导致其洗脱。然而,在三个剥离部分中的任一个中仅小部分装载的
211
at被洗脱,总洗脱率合计为约13%。意想不到的结果是,大多数
211
at(约79%)保持粘附在多孔珠上。虽然洗脱率低于理想值,但回收的
211
at产物具有高纯度,其对bi和其他放射化学污染物完全去污,且去污因子(df)≥105。
[0148]
目前,
211
at与树脂粘附的确切机理尚不清楚,但似乎存在ato
+
与树脂骨架的相互作用。树脂基于聚苯乙烯二乙烯苯聚合物。当用作吸附到表面的萃取剂配体薄膜的支撑物时,如金属离子的萃取色谱法,该骨架被认为对感兴趣的金属物种是惰性的。ato
+
物种保留在树脂上的一个原因可能是ato
+
分子与聚合物中缺陷的相互作用,这些缺陷是在树脂制造中产生的。
211
at与聚合物链中的缺陷相互作用而不是与树脂的整体官能度(在情况下为苯基)相互作用的假设,是保留非常少量的
211
at。也就是说,树脂保留了大约10μci的
211
at,换算为2.4
×
10-14
mol,比存在的苯基少多个数量级。如果苯基和
211
at发生强的相互作用,预期的结果将是
211
at具有接近定量的
保留。另一方面,如果与缺陷发生相互作用,这些缺陷只需要占树脂的很小部分,《《0.01%(w/w%)。
[0149]
随后,用少量掺料的也在2m hno3中的第1回溶解的轰击靶溶液(约13μci
211
at)对使用3-辛酮替代1-辛醇浸渍的第二萃取色谱法系统进行测试(见图5)。与1-辛醇相比,使用3-辛酮具有多个优点,其包括在较高的hno3浓度下显示出增强的
211
at萃取,并且在生物学上是无害的。与1-辛醇非常相似,3-辛酮不萃取bi,d值《0.1,而bi保留在装载溶液中通过柱。放射化学杂质也保留在装载溶液中,不受阻碍地通过柱。
211
at被3-辛酮体系更强地萃取(≥99%),在洗涤或冲洗中具有最小浸出(《3%)。
211
at在乙醇剥离液中的洗脱率提高到37%,在仅第二剥离液部分中就洗脱了近22%。尽管这提高了产率,但大多数(59%)的
211
at仍保留在树脂上。同样需要指出的是,尽管产率低,但产物纯度非常高,df≥105。与1-辛醇体系一样,少量的(约7.7μci或1.8
×
10-14
mol)
211
at保留在树脂上,符合聚合物链缺陷是保留来源的假设。
[0150]
在3-辛酮体系的洗脱率提高之后,将装载溶液的酸度增加到6m hno3(色谱图如图6所示),以确定是否需要在增加
211
at的量之前调整溶解的酸度。与在2m hno3中的装载一样,
211
at被萃取(≥98%),而bi和其他污染物没有被保留在柱上,而是随着装载溶液移动通过柱。
211
at的洗脱图与以2m hno3装载的洗脱图相当,在第二剥离部分中约有25%被洗脱,三个剥离部分总共为43%,具有高纯度(df≥105)。大部分
211
at留在树脂上,为53%(约6.9μci或1.6
×
10-14
mol),可能地结合在聚合物的缺陷位点。虽然洗脱率适中,但
211
at直到剥离才洗脱表明溶解的轰击靶溶液可以在不改进的情况下直接装载。
[0151]
然后,将掺料的活性增加到约1mci的
211
at,并检查了3-辛酮浸渍树脂的洗脱曲线作为硝酸浓度的函数。图7-9分别示出了
211
at在2、4和5.7m hno3装载中的色谱图。在这三种情况下(见表2),与以前的研究一样,
211
at在柱上的初始装载量接近定量(~97%),而bi和放射化学杂质再次不受阻碍地通过柱。任何残留的杂质在2m hno3洗涤和h2o冲洗中去除。然后大部分(71-77%)的
211
at在剥离中从柱中洗脱,其中40-44%在第一个部分中被洗脱,随后20-23%在第二个部分中被洗脱。与仅13μci 211
at的色谱图的一个主要区别是,随着
211
at量的增加,树脂似乎保留了更小百分比的装载的
211
at,仅19-24%,而不是以前研究中观察到的50-59%。应注意,在这些色谱图中,试图通过调整收集量约250μl来使部分收集与溶剂前端对齐。这似乎略高于估计,因为在死体积部分中观察到少量的
207
bi和
66/67
ga。
[0152]
表2:使用3-辛酮浸渍的cg300m树脂床(0.5ml bv,7mm id
×
13mm高)的含有399-μl第2回溶解的轰击靶掺料(~1.0mci 211
at)的2m hno3的1.5ml等分试样、4m hno3的1.4ml等分试样和5.7m hno3的1.3ml等分试样的色谱图的比较。
[0153][0154]
*注:死体积被假定为bv的一半,但似乎已经过高估计,因为在部分中观察到少量的
207
bi以及
66/67
ga;数据进行了衰变校正,以说明半衰期的差异。
[0155]
用第1回溶解的靶溶液(约6m hno3中含约41mci 211
at)的5ml等分试样进一步放大分离。图10示出了放大分离的洗脱图。与含有较少量
211
at的研究一样,柱中没有保留污染物物种,bi和其他不期望的物种在装载中一起被带走。即使有大得多的质量,bi也在洗涤液部分中完全从柱中移除。更引人感兴趣的是放大分离中的
211
at洗脱图。首先,
211
at(约4mci)的≥98%被装载到柱上,在整个5-ml的装载量中没有观察到突破。其次,
211
at在洗涤和冲洗过程中大约仅1%的渗漏。第三,大部分
211
at在前两个剥离部分中被洗脱,分别为32%和61%,占装载的
211
at的95%,df≥105。再次,应注意的事实是,对于该色谱图,没有试图调整部分收集以对应于装载溶液前端。因此,每个部分的前40-50%的溶液是前一部分的残余溶液,其保留在柱的自由体积中。随后的三个剥离部分含有最小量的
211
at,分别为1%、0.3%和≤0.1%。最后,树脂仅保留了极少量的
211
at,≤2%,其表示82μci(约1.9
×
10
13
mol)。
[0156]
最后,
211
at的量增加到约9.8mci,如图11所示。与4.1mci的
211
at分离中观察到的趋势一致,bi和其他放射化学污染物仍留在上清液中,而
211
at被柱强烈保留,其约95%装载量。在洗涤和冲洗中观察到很少(≤1%)的
211
at渗漏,直到剥离才被洗脱,其中大部分(约71%)在第一剥离部分中被回收。连续剥离部分包含约16%、3%、1.2%和≤1%的
211
at。剥离中所有回收的
211
at的df仍然很高,》105。在剥离的第一部分中的较大百分比是由于部分的收集被调整以适应柱的自由体积的事实。同样,少量的
211
at保持粘附在树脂上,约2%。当
211
at的量从约4.1mci(9.4
×
10-12
mol)增加到9.8mci(2.3
×
10-11
mol)时,
211
at的行为仍然非常相似,尽管总量增加了一倍以上-该因数为2.4x-211
at的实际量的增加仍然很小,为1.4
×
10-11
mol。这使得化学体系中的其他组分,特别是3-辛酮(2.3
×
10-3
mol),过量得多得多,大约9个数量级。考虑到这一点,正在研究的分离体系应该能够适应
211
at的进一步增加,并预计将非常适合于生产规模为20-100mci 211
at的轰击的靶。
[0157]
211
at的形态(在溶解液中的主要为hno3;在浸渍树脂中萃取的,主要为3-辛酮;在剥离中洗脱的,主要为乙醇)还没有实验测定,因为由半衰期短而导致的少量的at存在阻碍了传统的光谱技术的应用。at(iii)(作为ato
+
)是水溶液中的主要物种。计算了ato
+
的空π*
轨道与酮的o的
‘
sp2’
杂化孤对电子之间强的供体-受体相互作用,并认为这是ato
+
从hno3中有效萃取到3-辛酮中的驱动力。因此,ato
+
可能是乙醇剥离中的主要物种。
[0158]
尽管手工加入洗脱液和收集部分的性质很慢,但从加入第一个0.5-ml装载等分试样到洗脱第五个和最后一个剥离部分的整个过程花了不到20分钟。假设用泵将溶液送入通过柱,设定为2ml min-1
的中等速度,该回收≥95%的
211
at的过程将花费不到5分钟。如前所述,ato
+
和树脂之间的亲和力的来源目前未知,这阻碍了给出为什么放大分离的产率比较小分离的产率高得多的详细描述。然而,如果推测与树脂中的缺陷发生相互作用,则得出树脂上所有暴露的缺陷位点都在放大分离中饱和的结论并非不合理。一旦位点饱和,大部分
211
at被萃取到填充孔隙的溶剂中,通过将3-辛酮溶解到乙醇剥离液中来洗脱。这种直接从硝酸中以接近定量的产率回收高纯度
211
at的方法代表了at分离的重大进展。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1