基于MOFs的3D纳米印迹孔道及其合成方法与应用
基于mofs的3d纳米印迹孔道及其合成方法与应用
技术领域
1.本发明属于功能化催化剂用于高级氧化技术处理废水中难降解有机污染物的研究领域,具体涉及基于mofs的3d纳米印迹孔道及其合成方法与应用。
背景技术:
2.高级氧化技术(aops)是通过催化剂催化活化氧化剂产生活性氧物质(ros),通过ros的高氧化作用氧化降解污染物。被认为是一个很有前景的去除废水中的难降解有机污染物的水处理技术。但是由于氧化自由基半衰期短,如在aops水处理过程中通常使用的so4·-在的半衰期为30
–
40μs,
·
oh的半衰期为20 ns,且自由基的动力学传质速率一般在3-8
×
108m-1
s-1
,同时废水中大量污染物含量低,因此在降解反应中自由基与污染物的接触机会少,导致在废水深度处理过程中污染物的去除效率极低。
3.目前大量研究只聚焦于通过提高ros的产生率来提高污染物的去除率,这些研究可以实现污染物中c-x(x=cl、br等)键断裂、-oh、-ch3等支链从苯环上的分离及苯环和杂环化合物的开环,但基于反应基质的不同,难降解有机污染物的氧化过程矿化不完全,产生的副产物毒性呈现波动趋势,有的甚至比原始的污染物毒性更高。现有催化体系中催化剂能提供的催化活性位点较多,自由基产生释放速率过快,瞬间可产生大量自由基,如不能被充分利用就会被淬灭,从而降低自由基的利用效率,同时由于水中污染物的浓度都属于微量级,自由基氧化污染物过程中自由基的传输距离长,因此若采用现有催化体系催化活化氧化剂产生自由基的方式,大部分在利用前会被催化剂或反应体系中还原性物质淬灭,导致自由基的浪费。因此,在限域效应的作用下靶向去除污染物可以达到高效降解和提高自由基利用率的目的。
4.将金属有机骨架材料(metal organic frameworks,mofs)作为高级氧化技术的催化剂是近年的研究热点。mofs具有可调控的拓扑结构性质,能够通过选择合适的组装方法生成具有催化反应所需金属活性中心和几何拓扑结构的有序晶体,同时其不同的有机配体含氮杂环配体、羧酸类配体等结构,为mofs功能化修饰提供了良好的结构基础。
5.相关研究报道了在mofs表面构建一层分子印迹层,表面分子印迹催化剂将污染物吸附在印迹层表面,氧化剂需要穿过印迹层与催化活性中心作用产生自由基,使得氧化剂与催化中心的传质阻力增大,导致污染物的降解效率低(xitong li,jinquanwan,yanwang,haiyuan chi,zhicheng yan,su ding,selective removal and persulfate catalytic decomposition of diethyl phthalate from contaminated water on modified mil100 through surface molecular imprinting[j],chemosphere,240(2020)124875)。
技术实现要素:
[0006]
为了克服现有材料和技术的不足,本发明的目的在于提供基于mofs的3d纳米印迹孔道及其合成方法与应用,通过在mofs表面构建3d纳米孔道,将分子印迹技术引入纳米孔道中,使3d纳米印迹孔道可靶向吸附污染物并彻底降解。本发明采用前驱体法,采用具有3d
纳米孔道作为合成印迹位点的前驱体,然后将污染物特异性识别位点引入孔道中,模板分子与功能单体在孔道内壁上形成形状、官能团等结构互补的印迹腔,待模板分子洗脱之后形成的印迹腔可精准识别目标污染物。该3d纳米印迹孔道前驱体在mofs表面聚合形成核壳结构。由于识别位点在3d印迹孔道中分布,目标污染物与自由基之间的传质距离缩短,同时在纳米限域效应的作用下,污染物降解速率加快,污染物的去除效率大幅度提高。此外,自由基的利用率提高,处理成本降低,易于工程实用。
[0007]
本发明提供的一种基于mofs的3d纳米印迹孔道的合成方法,旨在解决难降解有机污染物在废水中降解效率低的问题。3d纳米印迹孔道(3d nano-imprinted channels,3d-nic)催化剂耦合了吸附和降解的功能,3d纳米印迹孔道具有三方面的作用:(1)在降解过程中,腐殖酸等大分子天然有机污染物会掩蔽催化剂的活性位点,导致污染物去除效率低。鉴于此,在3d-nics上构建了与目标污染物尺寸大小相似的传质孔道,通过孔道的特定尺寸,基于尺寸排阻效应,3d纳米印迹孔道可对目标污染物进行初步的筛选,避免非污染物物质对降解反应的影响;(2)为了实现低浓度污染物的富集,3d-nics通过特定识别位点的形状、成键特性和极性作用的记忆效应,可对目标污染物进一步精确的识别并靶向吸附在在印迹孔道中;(3)自由基与目标污染物的降解反应被局限在3d纳米印迹孔道中,在限域效应的作用下,印迹孔道内的自由基浓度增大且传质距离缩短,使自由基与目标污染物的接触机会增加,目标污染物及其产生的中间产物可彻底被自由基降解,从而达到污染物高效去除的目的。3d-nics在高级氧化技术(aops)的应用,可大大提高降解反应效率,提高自由基的利用率,实现污染物的深度降解。
[0008]
基于mofs的3d纳米印迹孔道的合成方法,包括步骤如下:
[0009]
(1)带氨基的fe基mofs催化中心供体的制备:将fecl3·
6h2o和2-氨基对苯二甲酸加入n,n-二甲基甲酰胺(dmf)溶剂中搅拌30-60min。将制备好的混合物转移到聚四氟乙烯内衬的高压釜反应器中,在100-150℃加热18-22h。产物分别用dmf、甲醇和水洗涤,纯化后真空干燥12-15h,所得产物为带氨基的fe基mofs催化中心供体。
[0010]
(2)3d纳米印迹孔道前驱液的制备:先将模板分子和3d孔道前驱体加入反应溶剂中混合均匀,然后加入带氨基的fe基mofs催化中心供体、功能单体和交联剂搅拌,充入惰性气体,静置8-16h使反应混合液进行预聚合,得到预聚物溶液。
[0011]
(3)前驱体法制备3d纳米印迹孔道:在步骤(2)所制备的预聚物溶液中加入引发剂,惰性气体氛围下保持20-60min排出溶液中的氧气,然后将混合液置于40-80℃的水浴中进行聚合反应,反应需在搅拌(转速为100-300r/min)下进行8-16h,得到聚合物。
[0012]
(4)基于mofs的3d纳米印迹孔道中模板分子的洗脱:将步骤(3)制备的聚合物进行离心分离,所得聚合物进行洗脱,采用甲醇/乙酸混合液(体积比为5:1-9:1)进行索氏萃取洗脱8-16h。然后用甲醇、水分别洗脱聚合物,待模板分子完全洗脱后离心分离,将所得聚合物在40-80℃的真空干燥箱中干燥8-16h,得到基于mofs的3d纳米印迹孔道。
[0013]
进一步地,步骤(1)中,所述fecl3·
6h2o的投加量为2.00-4.00mm;2-氨基对苯二甲酸的投加量为1.00-2.00mm;n,n-二甲基甲酰胺的投加量为20-50ml。
[0014]
进一步地,步骤(2)中,所述模板分子即目标污染物为磺胺甲恶唑(sulfamethoxazole,smx),所述3d孔道前驱体可为α-环糊精、β-环糊精、γ-环糊精、碳纳米管、分子筛等中的一种以上,所述功能单体为丙烯酸、丙烯腈、甲基丙烯酸等中的一种以上,
反应溶剂为二甲基亚砜、乙腈等中的一种以上,交联剂为正硅酸乙酯(四乙氧基硅烷)、二甲基丙烯酸乙二醇酯、二乙烯基苯、乙二醇二甲基丙烯酸酯等中的一种以上。
[0015]
进一步地,步骤(2)中,所述3d孔道前驱体与模板分子的质量比为2:1-8:1;所述功能单体与模板分子的质量比为2:5-4:5;所述功能单体与交联剂的质量比为1:1-1:3;所述3d孔道前驱体与带氨基的fe基mofs催化中心供体的质量比为5:1-20:1;反应溶剂体积为30-100ml。
[0016]
进一步地,步骤(2)中,所述3d孔道前驱体与模板分子(smx)的质量比为3:1-7:1。
[0017]
进一步地,步骤(2)中,所述3d孔道前驱体与模板分子(smx)的质量比为3:1-6:1。
[0018]
进一步地,步骤(2)中,所述功能单体与模板分子的质量比为3:5-4:5。
[0019]
进一步地,步骤(2)中,所述3d孔道前驱体与带氨基的fe基mofs催化中心供体的质量比为5:1-15:1。
[0020]
进一步地,步骤(2)中所述充入惰性气体的时间为20-40min。
[0021]
进一步地,步骤(2)中,预聚合静置时间为8-15h。
[0022]
进一步地,步骤(3)中,聚合反应的温度为50-80℃。
[0023]
进一步地,步骤(4)中,洗脱时间为8-15h。
[0024]
进一步地,步骤(4)中,真空干燥温度为50-80℃。
[0025]
进一步地,步骤(3)所述引发剂为偶氮二异丁腈,其添加量为0.02-0.06g。
[0026]
本发明提供所述合成方法制备的基于mofs的3d纳米印迹孔道。
[0027]
进一步地,mofs的3d纳米印迹孔道具有纳米限域效应、尺寸排阻效应和靶向识别效应。
[0028]
本发明还提供所述基于mofs的3d纳米印迹孔道应用于高级氧化技术中高效去除废水中的有机微污染物,将基于mofs的3d纳米印迹孔道加入含有有机微污染物的废水中,再加入氧化剂,置于恒温振荡培养箱中振荡反应,反应温度为20-40℃。
[0029]
进一步地,所述有机微污染物为磺胺类抗生素。
[0030]
进一步地,所述有机微污染物为磺胺甲恶唑(smx)。
[0031]
进一步地,所述氧化剂为过硫酸盐或h2o2。
[0032]
进一步地,所述高级氧化技术为过硫酸盐体系或h2o2体系。
[0033]
进一步地,所述有机微污染物和氧化剂的摩尔比为50:1-350:1,基于mofs的3d纳米印迹孔道在废水中的浓度为0.1g/l-1g/l,有机微污染物在废水中的浓度为5mg/l-45mg/l。
[0034]
本发明针对废水深度处理过程中有机微污染物难去除的问题,公开了一种基于mofs的3d纳米印迹孔道催化剂应用于高级氧化技术,本发明提供的基于mofs的3d纳米印迹孔道催化剂具有以下优点:
[0035]
(1)本发明提出的一种基于mofs的3d纳米印迹孔道的合成方法设备简单,反应条件温和,过程易于控制,无需添加有毒有害试剂,便于催化剂的产业化。
[0036]
(2)本发明提供的前驱体法所使用的3d孔道前驱体可选择性强,提高了本发明所合成的基于mofs的3d纳米印迹孔道形成的可行性。
[0037]
(3)本发明提供基于mofs的3d纳米印迹孔道催化剂可根据孔道尺寸大小对污染物进行初步识别,然后通过印迹腔的形状、大小和价键作用精准识别目标污染物,实现目标污
染物的靶向去除。
[0038]
(4)本发明的基于mofs的3d纳米印迹孔道催化剂将印迹腔置于纳米孔道中,可避免废水中干扰组分对识别位点的掩蔽,同时纳米印迹孔道可发挥限域效应的作用,加速污染物降解反应,提高污染物降解效率和自由基利用率,降低处理成本。
[0039]
(5)本发明提供的基于mofs的3d纳米印迹孔道的合成方法是在mofs基底的表面构建印迹孔道层,该合成方法可确保催化剂长期使用的稳定性,有利于3d纳米印迹孔道催化剂的实际应用。
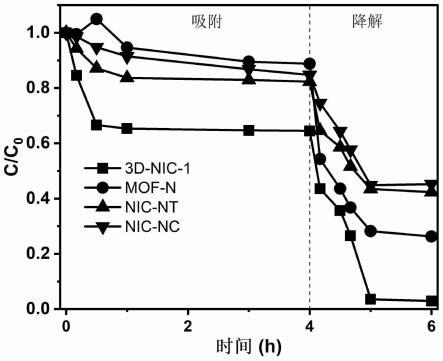
附图说明
[0040]
图1为基于mofs的3d纳米印迹孔道催化剂结构及靶向降解示意图。
[0041]
图2为本发明实施例1基于mofs的3d纳米印迹孔道催化剂的x射线衍射(xrd)谱图。
[0042]
图3为本发明应用例1基于mofs的3d纳米印迹孔道催化剂在混合水体中对smx的靶向吸附容量图。
[0043]
图4为本发明应用例2基于mofs的3d纳米印迹孔道对废水中smx的靶向吸附及降解效率图。
具体实施方式
[0044]
为使本公开的合成方法、技术可行性和优点更加清楚明白,以下结合附图对本发明的具体实施作进一步的详细说明,但本发明的实施和保护不限于此。
[0045]
实施例1
[0046]
本实施例提供了一种基于mofs的3d纳米印迹孔道的合成方法,主要步骤如下:
[0047]
(1)带氨基的fe基mofs催化中心供体的合成:称取0.675g的fecl36
·
h2o(2.50mmol)和0.225g的2-氨基对苯二甲酸(1.25mmol)溶解在30ml的n,n-二甲基甲酰胺(dmf)溶液中,连续搅拌30min。将上述混合物转移到一个100ml的聚四氟乙烯内衬的高压反应釜中,在110℃加热20h。产物分别用dmf、甲醇和去离子水洗涤三次,离心分离后在60℃真空下干燥12h,所制备的材料命名为mof-n。
[0048]
(2)3d-nic催化剂的合成:称取0.50g磺胺甲恶唑(smx)作为模板分子,加入2.5g的β-环糊精(β-cd)作为3d孔道前驱体,加入50ml的二甲亚砜(dmso)中搅拌混合均匀,然后加入0.20g步骤(1)所制备的mof-n,1.31ml(0.30g)甲基丙烯酸(maa)和0.60g的乙二醇二甲基丙烯酸酯(egdma),搅拌30min,通入n230 min已排除氧气,静置12h形成预聚物溶液。
[0049]
(3)在步骤(2)的预聚物溶液中加入0.05g偶氮二异丁腈(aibn),通入n2净化40min后置于60℃水浴中以转速为200r/min搅拌反应12h,到反应完全,离心分离,得到聚合物,将聚合物用甲醇/醋酸(v/v=7:1)混合溶剂索氏萃取法洗脱12h,使聚合物中模板分子与印迹腔的成键断裂。再分别用无水甲醇和超纯水各洗涤3次使模板分子洗脱干净,离心分离,所得材料在60℃下干燥12h,得到基于mofs的3d纳米印迹孔道,命名为3d-nic-1,其结构如图1所示,由图1可知在催化中心mof-n表面构建了印迹孔道层,污染物被吸附在印迹孔道中,催化中心活化氧化剂产生自由基,自由基迅速攻击被吸附在孔道中的污染物,使污染物高效降解。
[0050]
实施例2
[0051]
(1)带氨基的fe基mofs催化中心供体的合成:称取0.675g的fecl36
·
h2o(2.50mmol)和0.225g的2-氨基对苯二甲酸(1.25mmol)溶解在30ml的n,n-二甲基甲酰胺(dmf)溶液中,连续搅拌30min。将上述混合物转移到一个100ml的聚四氟乙烯内衬的高压反应釜中,在110℃加热20h。产物分别用dmf、甲醇和去离子水洗涤三次,离心分离后在60℃真空下干燥12h,所制备的材料命名为mof-n。
[0052]
(2)3d-nic催化剂的合成:称取0.50g磺胺甲恶唑(smx)作为模板分子,加入3.5g的β-环糊精(β-cd)作为3d孔道前驱体,加入50ml的二甲亚砜(dmso)中搅拌混合均匀,然后加入0.20g步骤(1)所制备的mof-n,0.4g甲基丙烯酸(maa)和1.2g的乙二醇二甲基丙烯酸酯(egdma),搅拌30min,通入n230 min已排除氧气,静置12h形成预聚物溶液。
[0053]
(3)在步骤(2)的预聚物溶液中加入0.05g偶氮二异丁腈(aibn),通入n2净化40min后置于60℃水浴中以转速为200r/min搅拌反应12h,到反应完全,离心分离,得到聚合物,将聚合物用甲醇/醋酸(v/v=9:1)混合溶剂索氏萃取法洗脱12h,使聚合物中模板分子与印迹腔的成键断裂。再分别用无水甲醇和超纯水各洗涤3次使模板分子洗脱干净,离心分离,所得材料在60℃下干燥12h,得到基于mofs的3d纳米印迹孔道,命名为3d-nic-2。
[0054]
实施例3
[0055]
(1)带氨基的fe基mofs催化中心供体的合成:称取0.675g的fecl36
·
h2o(2.50mmol)和0.225g的2-氨基对苯二甲酸(1.25mmol)溶解在30ml的n,n-二甲基甲酰胺(dmf)溶液中,连续搅拌30min。将上述混合物转移到一个100ml的聚四氟乙烯内衬的高压反应釜中,在110℃加热20h。产物分别用dmf、甲醇和去离子水洗涤三次,离心分离后在60℃真空下干燥12h,所制备的材料命名为mof-n。
[0056]
(2)3d-nic催化剂的合成:称取0.50g磺胺甲恶唑(smx)作为模板分子,加入如表1所述用量的3d孔道前驱体(β-环糊精(β-cd)),步骤(1)所制备的mof-n,甲基丙烯酸(maa),mof-n,乙二醇二甲基丙烯酸酯(egdma)于50ml的二甲亚砜(dmso)中搅拌30min混合均匀,通入n230 min已排除氧气,静置12h形成预聚物溶液。
[0057]
(3)在步骤(2)的预聚物溶液中加入0.05g偶氮二异丁腈(aibn),通入n2净化40min后置于60℃水浴中以转速为200r/min搅拌反应12h,到反应完全,离心分离,得到聚合物,将聚合物用甲醇/醋酸(v/v=7:1)混合溶剂索氏萃取法洗脱12h,使聚合物中模板分子与印迹腔的成键断裂。再分别用无水甲醇和超纯水各洗涤3次使模板分子洗脱干净,离心分离,所得材料在60℃下干燥12h,得到基于mofs的3d纳米印迹孔道。
[0058]
基于mofs的3d纳米印迹孔道的合成条件,详细的合成控制条件如下表1所示
[0059][0060]
实施例4
[0061]
(1)带氨基的fe基mofs催化中心供体的合成:称取0.675g的fecl36
·
h2o(2.50mmol)和0.225g的2-氨基对苯二甲酸(1.25mmol)溶解在30ml的n,n-二甲基甲酰胺(dmf)溶液中,连续搅拌30min。将上述混合物转移到一个100ml的聚四氟乙烯内衬的高压反应釜中,在110℃加热20h。产物分别用dmf、甲醇和去离子水洗涤三次,离心分离后在60℃真空下干燥12h,所制备的材料命名为mof-n。
[0062]
(2)3d-nic催化剂的合成:称取0.50g磺胺甲恶唑(smx)作为模板分子,加入3d孔道前驱体(β-环糊精(β-cd))1.0g,步骤(1)所制备的mof-n 0.05g,甲基丙烯酸(maa)0.4g,乙二醇二甲基丙烯酸酯(egdma)0.4g于50ml的二甲亚砜(dmso)中搅拌40min混合均匀,通入n220 min已排除氧气,静置8h形成预聚物溶液。
[0063]
(3)在步骤(2)的预聚物溶液中加入0.05g偶氮二异丁腈(aibn),通入n2净化20min后置于80℃水浴中以转速为100r/min搅拌反应8h,到反应完全,离心分离,得到聚合物,将聚合物用甲醇/醋酸(v/v=7:1)混合溶剂索氏萃取法洗脱16h,使聚合物中模板分子与印迹腔的成键断裂。再分别用无水甲醇和超纯水各洗涤3次使模板分子洗脱干净,离心分离,所得材料在80℃下干燥16h,得到基于mofs的3d纳米印迹孔道。
[0064]
实施例5
[0065]
(1)带氨基的fe基mofs催化中心供体的合成:称取0.675g的fecl36
·
h2o(2.50mmol)和0.225g的2-氨基对苯二甲酸(1.25mmol)溶解在30ml的n,n-二甲基甲酰胺(dmf)溶液中,连续搅拌30min。将上述混合物转移到一个100ml的聚四氟乙烯内衬的高压反应釜中,在110℃加热20h。产物分别用dmf、甲醇和去离子水洗涤三次,离心分离后在60℃真空下干燥12h,所制备的材料命名为mof-n。
[0066]
(2)3d-nic催化剂的合成:称取0.50g磺胺甲恶唑(smx)作为模板分子,加3d孔道前驱体(β-环糊精(β-cd))4.0g,步骤(1)所制备的mof-n 0.8g,甲基丙烯酸(maa)0.3g,乙二醇二甲基丙烯酸酯(egdma)0.3g于50ml的二甲亚砜(dmso)中搅拌40min混合均匀,通入n240 min已排除氧气,静置16h形成预聚物溶液。
[0067]
(3)在步骤(2)的预聚物溶液中加入0.05g偶氮二异丁腈(aibn),通入n2净60min后
置于40℃水浴中以转速为300r/min搅拌反应16h,到反应完全,离心分离,得到聚合物,将聚合物用甲醇/醋酸(v/v=7:1)混合溶剂索氏萃取法洗脱8h,使聚合物中模板分子与印迹腔的成键断裂。再分别用无水甲醇和超纯水各洗涤3次使模板分子洗脱干净,离心分离,所得材料在40℃下干燥8h,得到基于mofs的3d纳米印迹孔道。
[0068]
对比例1
[0069]
(1)带氨基的fe基mofs催化中心供体的合成:称取0.675g的fecl36
·
h2o(2.50mmol)和0.225g的2-氨基对苯二甲酸(1.25mmol)溶解在30ml的n,n-二甲基甲酰胺(dmf)溶液中,连续搅拌30min。将上述混合物转移到一个100ml的聚四氟乙烯内衬的高压反应釜中,在110℃加热20h。产物分别用dmf、甲醇和去离子水洗涤三次,离心分离后在60℃真空下干燥12h,所制备的材料命名为mof-n。
[0070]
(2)nic-nt催化剂的合成:不加入模板分子(smx),直接在50ml的二甲亚砜(dmso)中加入2.5g的β-环糊精(β-cd)作为3d孔道前驱体搅拌混合均匀,然后加入0.20g步骤(1)所制备的mof-n,1.31ml(0.30g)甲基丙烯酸(maa)和0.60g的乙二醇二甲基丙烯酸酯(egdma),搅拌30min,通入n230 min已排除氧气,静置12h形成预聚物溶液。
[0071]
(3)在步骤(2)的预聚物溶液中加入0.05g偶氮二异丁腈(aibn),通入n2净化40min后置于60℃水浴中以转速为200r/min搅拌反应12h,到反应完全,离心分离,得到聚合物,将聚合物用甲醇/醋酸(v/v=7:1)混合溶剂索氏萃取法洗脱12h,使聚合物中模板分子与印迹腔的成键断裂。再分别用无水甲醇和超纯水各洗涤3次使模板分子洗脱干净,离心分离,所得材料在60℃下干燥12h,合成条件控制如表2所示,得到基于mofs的3d非印迹纳米孔道,命名为nic-nt。
[0072]
对比例2
[0073]
(1)带氨基的fe基mofs催化中心供体的合成:称取0.675g的fecl36
·
h2o(2.50mmol)和0.225g的2-氨基对苯二甲酸(1.25mmol)溶解在30ml的n,n-二甲基甲酰胺(dmf)溶液中,连续搅拌30min。将上述混合物转移到一个100ml的聚四氟乙烯内衬的高压反应釜中,在110℃加热20h。产物分别用dmf、甲醇和去离子水洗涤三次,离心分离后在60℃真空下干燥12h,所制备的材料命名为mof-n。
[0074]
(2)nic-nc催化剂的合成:不加入3d孔道前驱体(β-环糊精(β-cd)),称取0.50g磺胺甲恶唑(smx)作为模板分子在50ml的二甲亚砜(dmso)中搅拌混合均匀,然后加入0.20g步骤(1)所制备的mof-n,1.31ml(0.30g)甲基丙烯酸(maa)和0.60g的乙二醇二甲基丙烯酸酯(egdma),搅拌30min,通入n230 min已排除氧气,静置12h形成预聚物溶液。
[0075]
(3)在步骤(2)的预聚物溶液中加入0.05g偶氮二异丁腈(aibn),通入n2净化40min后置于60℃水浴中以转速为200r/min搅拌反应12h,到反应完全,离心分离,得到聚合物,将聚合物用甲醇/醋酸(v/v=7:1)混合溶剂索氏萃取法洗脱12h,使聚合物中模板分子与印迹腔的成键断裂。再分别用无水甲醇和超纯水各洗涤3次使模板分子洗脱干净,离心分离,所得材料在60℃下干燥12h,合成条件控制如表2所示,得到基于mofs的3d印迹材料,命名为nic-nc。
[0076]
基于mofs的3d印迹催化剂(nic-nc)和基于mofs的3d非印迹纳米孔道催化剂(nic-nt)的合成原料如表2所示,其余合成步骤同实施例1。
[0077]
表2基于mofs的3d非印迹纳米孔道催化剂和基于mofs的3d印迹催化剂的合成原
料。
[0078][0079]
检测实施例1制备的基于mofs的3d纳米印迹孔道是否合成成功,如图2所示,mof-n和β-cd具有不同的晶面峰,说明两种物质的晶体结构不同。实施例1制备的3d-nic-1包含β-cd和mof-n的峰,表明3d-nic-1中具有β-cd的孔道结构,同时保留了催化中心mof-n的晶体结构。说明3d-nic-1中形成了纳米印迹孔道。
[0080]
应用例1
[0081]
应用例1比较纳米孔道和印迹腔在实施例1制备的3d-nic-1催化剂的尺寸排阻效应和靶向吸附性能。
[0082]
选取不同结构的污染物即磺胺甲恶唑(smx)、橙黄g(og)、环丙沙星(cip)和四环素(tet)配制混合溶液(溶剂为水),探索实施例1制备的3d-nic-1的尺寸排阻效应和靶向吸附性能。各污染物的浓度均为20mg/l,在100ml的上述混合溶液中分别加入0.05g的3d-nic-1、mof-n、nic-nt和nic-nc催化剂,将反应溶液置于恒温振荡培养箱中,转速控制在180rpm,温度为25℃。在吸附平衡后(12h)进行取样检测催化剂的对不同污染物的吸附容量。
[0083]
上述四种催化剂对不同污染物的吸附容量如图3所示,3d-nic对目标污染物smx的吸附容量可达80mg/g,而mof-n、nic-nt和nic-nc对smx吸附容量分别为33.5mg/g、25.6mg/g和44.8mg/g,说明本发明实施例1制备的3d纳米印迹孔道具有良好的吸附性能;mofs对污染物的吸附性能相当,而nic-nc对目标污染物表现出优先吸附趋势。选择性吸附实验表明,本发明实施例1制备的3d-nic-1在混合水样中对目标污染物smx表现出选择性且吸附容量最高,nic-nc吸附容量次之,说明分子印迹技术可在废水中通过印迹腔的大小、形状和价键作用吸附目标污染物。
[0084]
应用例2
[0085]
应用例2比较纳米孔道和印迹腔在实施例1制备的3d-nic-1催化剂的尺寸排阻效应和靶向吸附性能。
[0086]
选取不同结构的污染物即磺胺甲恶唑(smx)、橙黄g(og)、环丙沙星(cip)和四环素(tet)配制混合溶液(溶剂为水),探索实施例1制备的3d-nic-1的靶向吸附性能。各污染物的浓度均为45mg/l,在100ml的上述混合溶液中分别加入0.01g的3d-nic-1、mof-n、nic-nt和nic-nc催化剂,将反应溶液置于恒温振荡培养箱中,转速控制在180rpm,温度为25℃。在吸附平衡后(12h)进行取样检测催化剂的对不同污染物的吸附容量。
[0087]
应用例3
[0088]
应用例3比较纳米孔道和印迹腔在实施例1制备的3d-nic-1催化剂的尺寸排阻效应和靶向吸附性能。
[0089]
选取不同结构的污染物即磺胺甲恶唑(smx)、橙黄g(og)、环丙沙星(cip)和四环素(tet)配制混合溶液(溶剂为水),探索实施例1制备的3d-nic-1的尺寸排阻效应和靶向吸附
性能。各污染物的浓度均为5mg/l,在100ml的上述混合溶液中分别加入0.1g的3d-nic-1、mof-n、nic-nt和nic-nc催化剂,将反应溶液置于恒温振荡培养箱中,转速控制在180rpm,温度为25℃。在吸附平衡后(12h)进行取样检测催化剂的对不同污染物的吸附容量。
[0090]
应用例4
[0091]
应用例4证实本发明实施例1制备的3d-nic-1催化剂的限域效应对提高污染物降解性能的影响。
[0092]
将0.05g的mof-n、3d-nic、nic-nt和nic-nc四种催化剂分别加入盛有100ml 25mg/l smx溶液(溶剂为水)的反应瓶中,将反应瓶置于恒温振荡培养箱中,转速控制在180rpm,温度25℃。在吸附平衡后(4h)进行取样检测污染物的浓度。然后向四种反应溶液中加入过硫酸钠(ps)氧化剂,污染物smx与ps的摩尔比为200:1,待反应在设定的时间点(0min,5min,10min,20min,30min,1h,1.5h,2h,2.5h,3h,3.5h,4h)进行取样检测smx的浓度。
[0093]
上述四种材料对目标污染物的降解情况如图4所示,与其他三种材料相比,本发明实施例1制备的3d-nic-1的吸附性能更强。3d-nic、mof-n、nic-nt和nicn-c对smx的去除分别约为97%、60%、67%和66%。表明分子印迹技术将smx富集到纳米印迹孔道中,然后在限域效应的作用下使催化中心活化ps产生的自由基快速与smx反应,增加了smx与自由基的碰撞机率,加速smx的降解速率,自由基的利用率提高。
[0094]
本发明提供的基于mofs的3d纳米印迹孔道催化剂对复杂水体中低浓度(1-10mg/l)有机微污染物表现出高效降解率,基于mofs的3d纳米印迹孔道的形成可排除水体中腐殖酸、无机离子对催化活性中心和识别位点的掩蔽,实现废水深度处理的实际应用。基于mofs的3d纳米印迹孔道的限域效应可提高自由基的使用率,大大减少氧化剂的使用量,降低废水处理成本,具有很好的工程应用潜力。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1