一种碳包覆的负载型铂族单原子贵金属催化剂及其制备方法与应用
1.本发明属于催化剂技术领域,尤其涉及一种碳包覆的负载型铂族单原子贵金属催化剂及其制备方法与应用。
背景技术:
2.铈(ce)基氧化物负载贵金属催化剂在汽车尾气净化中具有广泛应用,其活性组分主要是由pt、pd和rh贵金属所组成。但现行商业化三效催化剂(ce基氧化物负载的pd-rh催化剂)仍面临如何提高低温冷启动活性(如150℃下co完全转化),增强其高温热稳定性并降低贵金属用量等技术挑战。由于贵金属价格居高不下,传统的贵金属颗粒催化剂的贵金属利用率低,而通过铂族金属的原子级分散能提高贵金属利用率,开发低贵金属用量的铂族金属基三效催化剂具有重大的应用需求。同时,三效催化剂在实际应用过程中面临高温水热、贫/富氧频繁切换等极端工况,高分散贵金属粒子不可避免地也会熟化-团聚-长大,从而导致催化剂失活。对于金属氧化物(如ceo2)负载的原子级分散金属催化剂,金属活性中心与载体主要是占据金属阳离子空位,通过形成金属-氧键(m-o键)来锚定金属活性中心,并形成以金属单原子为中心、包含临近氧的活性区域,而与金属原子配位的氧物种不仅起到锚定金属位点的作用,而且也具有极高的反应活性。但是在高温、还原气氛下,因其高反应活性极易与还原性气体分子(如co及h2等)反应,造成金属原子的锚定点(配体)缺少,使得金属原子极易发生团聚而形成颗粒,造成催化剂失活。因此如何开发原子级分散金属物种的稳定策略是开发高效、低成本汽车尾气催化剂的关键。
技术实现要素:
3.为此,本发明所要解决的技术问题在于克服现有技术中催化剂中贵金属利用率低,而高分散贵金属催化剂低温下活性差、高温下易失活的问题。
4.为解决上述技术问题,本发明提供了一种碳包覆的负载型铂族单原子贵金属催化剂及其制备方法与应用。
5.本发明的第一个目的是提供一种碳包覆的负载型铂族单原子贵金属催化剂的制备方法,包括以下步骤,
6.s1、将铂族贵金属盐溶液加入氧化铈载体溶液,经洗涤、干燥,得到氧化铈负载原子级铂族单原子贵金属催化剂,简记为m1/ceo2;
7.s2、将s1所述的氧化铈负载原子级铂族单原子贵金属催化剂分散于溶剂,调节分散液ph至7-9,加入儿茶酚胺进行反应,后经洗涤、干燥、热处理,得到所述碳包覆的负载型铂族单原子贵金属催化剂,简记为(m1/ceo2)@c。
8.在本发明的一个实施例中,在s1中,所述铂族贵金属盐溶液是将铂族贵金属盐溶于溶剂;所述溶剂为水、乙醇、乙二醇或dmf。
9.在本发明的一个实施例中,在s1中,所述铂族贵金属为钯、铂、铑、铱或钌。
10.在本发明的一个实施例中,在s1中,所述铂族贵金属盐溶液的浓度为0.01-0.50mmol/l,该浓度有利于贵金属在载体表面呈原子级分散状态。
11.在本发明的一个实施例中,在s1中,所述干燥的温度为60-80℃,时间为12-16h。
12.在本发明的一个实施例中,在s2中,所述儿茶酚胺为多巴胺、去甲基肾上腺素或左旋多巴。
13.在本发明的一个实施例中,在s2中,所述氧化铈负载原子级铂族单原子贵金属催化剂和儿茶酚胺的质量比为2:1-200:1。
14.进一步地,在s2中,所述氧化铈负载原子级铂族单原子贵金属催化剂和儿茶酚胺的质量比为5:1-40:1。通过改变儿茶酚胺的用量,可以增强碳包覆的负载型铂族单原子贵金属催化剂的低温活性。
15.在本发明的一个实施例中,在s2中,所述热处理的升温速率为2℃/min-5℃/min;温度为500-800℃,时间为2-5h。该升温速率有利于在铂族单原子贵金属催化剂表面形成均匀的碳包覆层。该温度下有利于去除来自儿茶酚胺的羟基基团。
16.在本发明的一个实施例中,在s2中,所述热处理是在惰性气氛下进行的。
17.在本发明的一个实施例中,在s2中,所述溶剂为水、乙醇、乙二醇或dmf。
18.在本发明的一个实施例中,在s2中,所述干燥的温度为40-80℃,时间为8-16h。
19.本发明的第二个目的是提供一种所述方法制备的碳包覆的负载型铂族单原子贵金属催化剂。
20.在本发明的一个实施例中,所述的碳包覆的负载型铂族单原子贵金属催化剂在还原气氛下热处理后仍保持很好的热稳定性。
21.在本发明的一个实施例中,还原气氛下热处理的升温速率为5℃/min-10℃/min;温度为300-500℃,时间为0.5-1h。
22.在本发明的一个实施例中,还原气氛为氢气和氩气的混合气氛,混合气氛中氢气的体积占比为5%-10%。
23.本发明的第三个目的是提供一种所述的碳包覆的负载型铂族单原子贵金属催化剂在催化co氧化反应中的应用。
24.在本发明的一个实施例中,所述应用中空速为15000-18000ml
·
g-1
·
h-1
,反应压力为0.1mpa。
25.在本发明的一个实施例中,所述应用中co的浓度为0.1-2vol.%,o2的浓度为4-20vol.%。
26.本发明的技术方案相比现有技术具有以下优点:
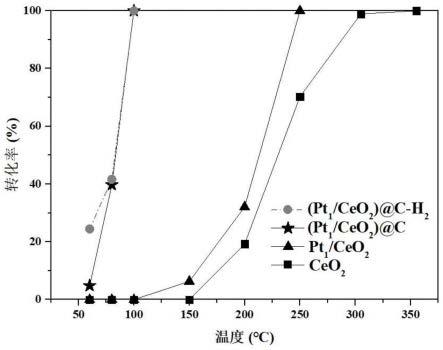
27.(1)本发明所述的碳包覆的负载型铂族单原子贵金属催化剂具有低温活性的原理是催化剂具有独特的m
1-o
2-n2活性位点,反应中o2和co分子吸附在m
1-o
2-n2位点上,其吸附能相较于在铂族金属表面的吸附能更弱,避免了co对原子级分散铂族金属的毒化。当o2吸附时,一个配位氧原子迁移到附近的表面氧空位,与此同时,被吸附的端部o2会桥接铂族金属和ce位点形成m-oo-ce构型。迁移的配位氧原子可以直接与被吸附的co结合形成co2分子,co2分子容易解吸,氧空位恢复。之后,o2在m
1-n2三角形位点上零活化能解离,一个分裂的氧原子补充m
1-o
2-n2构型中消耗的配位氧;另一个氧原子再次填补附近的表面氧空位,作为活性晶格氧直接将co氧化为co2,催化剂表面结构复原。
28.(2)本发明所述的碳包覆的负载型铂族单原子贵金属催化剂具有高温稳定性的原理是采用构建金属-非氧键的策略,通过微结构调控,使得原有ceo2负载的原子级分散金属中心配位结构由m
1-o4转变为m
1-o
2-n2,抑制还原性气体分子(如co,h2)与金属中心的配体相反应,从而提升原子级分散金属中心的热稳定性,使得金属活性中心在还原气氛下,保持原有的原子级分散状态,从而提高了催化剂热稳定性。
29.(3)本发明所述的碳包覆的负载型铂族单原子贵金属催化剂在空速为15000ml
·
g-1
·
h-1
,1vol.%co+20vol.%o2+n2的反应条件下,100℃下实现co的完全氧化,完成了150℃下co完全转化的技术挑战;且经高温h2预处理后,催化剂对co完全转化温度保持不变且金属活性中心仍保持100%分散度。
30.(4)本发明所述的碳包覆的负载型铂族单原子贵金属催化剂以开发贵金属利用率高的铂族单原子贵金属催化剂为技术方案,既提升了催化剂的低温活性,又提升了其高温热稳定性。
附图说明
31.为了使本发明的内容更容易被清楚地理解,下面根据本发明的具体实施例并结合附图,对本发明作进一步详细的说明,其中:
32.图1为本发明实施例1的(pt1/ceo2)@c的原子级倍率电镜图。
33.图2为本发明实施例1的(pt1/ceo2)@c的结构图,虚线圆圈内为催化剂的pt
1-o
2-n2活性位点。
34.图3为本发明实施例1的(pt1/ceo2)@c-h2、(pt1/ceo2)@c、pt1/ceo2和ceo2催化co氧化的反应性能图。
35.图4为本发明不同比例的pt1/ceo2和儿茶酚胺制备的(pt1/ceo2)@c、pt1/ceo2和ceo2催化co氧化的反应性能图。
36.图5为本发明实施例1的(pt1/ceo2)@c、pt1/ceo2、ceo2和对比例的(ceo2)@c催化co氧化的反应性能图。
具体实施方式
37.下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
38.实施例1
39.一种碳包覆的负载型铂族单原子贵金属催化剂及其制备方法与应用,具体包括以下步骤:
40.在25℃下,将0.013mmol氯铂酸加至50ml水中,制备成0.26mmol/l的金属前驱体溶液。将500mg氧化铈加至100ml超纯水(18.2mω)中,形成氧化铈载体分散液,使用蠕动泵将上述金属前驱体溶液逐滴滴加至氧化铈载体分散液中,之后,离心混合溶液并洗涤得到的固体,在60℃下干燥12h,得到氧化铈负载原子级分散pt催化剂pt1/ceo2。将上述pt1/ceo2分散在100ml超纯水(18.2mω)中,超声振动使pt1/ceo2均匀分散,调节分散液ph为8.5,加入质量为pt1/ceo2质量1/20的多巴胺,搅拌2h后离心混合溶液并洗涤得到的固体,在60℃下干燥12h,之后,在ar气氛下以5℃/min的升温速率升至600℃保持2h,得到碳包覆的氧化铈负载
原子级分散pt催化剂(pt1/ceo2)@c。将上述(pt1/ceo2)@c在5%h2/ar气氛下以10℃/min的升温速率升至400℃保持1h,得到经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂(pt1/ceo2)@c-h2。
41.haadf-stem拍摄的(pt1/ceo2)@c的原子级倍率电镜图如图1所示,白色圆圈内的亮点为负载的原子级分散的pt物种,白色虚线之间为惰性气氛处理后形成的包覆层。每个pt原子形成独特的pt
1-o
2-n2活性位点,都参与催化co氧化过程,提高了催化剂的低温活性和贵金属的利用率。
42.(pt1/ceo2)@c的结构如图2所示,白色圆圈内为pt
1-o
2-n2活性位点。反应过程中,o2吸附在pt
1-o
2-n2活性位点上,会桥接pt和ce形成pt-oo-ce构型,而配位氧原子迁移到附近的表面氧空位,与被吸附的co结合形成co2分子,co2分子解吸重新形成氧空位。之后,o2在pt
1-n2三角形位点上零活化能解离,一个分裂的氧原子补充pt
1-o
2-n2构型中消耗的配位氧原子;另一个氧原子再次填补表面氧空位,作为活性晶格氧直接将co氧化为co2,催化剂表面结构复原。
43.实施例2
44.基本同实施例1,将氯铂酸更换为硝酸钯,得到碳包覆的氧化铈负载原子级分散pd催化剂和经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pd催化剂。
45.实施例3
46.基本同实施例1,将氯铂酸更换为硝酸铑,得到碳包覆的氧化铈负载原子级分散rh催化剂和经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散rh催化剂。
47.实施例4
48.基本同实施例1,将氯铂酸更换为三氯化铱,得到碳包覆的氧化铈负载原子级分散ir催化剂和经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散ir催化剂。
49.实施例5
50.基本同实施例1,将氯铂酸更换为三氯化钌,得到碳包覆的氧化铈负载原子级分散ru催化剂和经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散ru催化剂。
51.实施例6
52.基本同实施例1,将多巴胺更换为去甲基肾上腺素,得到碳包覆的氧化铈负载原子级分散pt催化剂和经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂。
53.实施例7
54.基本同实施例1,将多巴胺更换为左旋多巴,得到碳包覆的氧化铈负载原子级分散pt催化剂和经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂。
55.实施例8
56.基本同实施例1,将多巴胺加入量更换为pt1/ceo2质量的1/40,得到碳包覆的氧化铈负载原子级分散pt催化剂和经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂。
57.实施例9
58.基本同实施例1,将多巴胺加入量更换为pt1/ceo2质量的1/10,得到碳包覆的氧化铈负载原子级分散pt催化剂和经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂。
59.实施例10
60.基本同实施例1,将多巴胺加入量更换为pt1/ceo2质量的1/5,得到碳包覆的氧化铈负载原子级分散pt催化剂和经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂。
61.实施例11
62.基本同实施例1,将h2气氛下的热处理温度更换为300℃,得到碳包覆的氧化铈负载原子级分散pt催化剂和经300℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂。
63.实施例12
64.基本同实施例1,将h2气氛下的热处理温度更换为500℃,得到碳包覆的氧化铈负载原子级分散pt催化剂和经500℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂。
65.应用例1
66.所有催化测试都是在固定床反应器中进行。分别将50mg实施例1的(pt1/ceo2)@c-h2、(pt1/ceo2)@c、pt1/ceo2和ceo2与500mg石英砂混合装入固定床反应器,以15000ml
·
g-1
·
h-1
的空速通入比例为1vol.%co+20vol.%o2+n2的气体,根据升温程序升高温度进行反应。通过带有甲烷转化炉的气相色谱仪(agilent gc-2060)分析反应物(co)和产物(co2),计算co转化率,结果如图3所示。从图3可以看出,(pt1/ceo2)@c和(pt1/ceo2)@c-h2在100℃下催化co完全转化。(pt1/ceo2)@c和(pt1/ceo2)@c-h2对于催化co氧化具有很好的低温活性和热稳定性,通过独特的pt
1-o
2-n2结构,稳定了与金属原子配位的氧物种,有效阻止了原子级分散pt贵金属物种在高温、还原气氛下的团聚,避免了催化剂活性的下降,在催化汽车尾气处理的应用中呈现出显著优势。
67.应用例2
68.基本同应用例1,分别将50mg实施例2制备的碳包覆的氧化铈负载原子级分散pd催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pd催化剂与500mg石英砂混合装入固定床反应器,都以15000ml
·
g-1
·
h-1
的空速通入比例为1vol.%co+20vol.%o2+n2的气体,根据升温程序升高温度进行反应。碳包覆的氧化铈负载原子级分散pd催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pd催化剂均在145℃下催化co完全转化。
69.应用例3
70.基本同应用例1,分别将50mg实施例3制备的碳包覆的氧化铈负载原子级分散rh催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散rh催化剂与500mg石英砂混合装入固定床反应器,都以15000ml
·
g-1
·
h-1
的空速通入比例为1vol.%co+20vol.%o2+n2的气体,根据升温程序升高温度进行反应。碳包覆的氧化铈负载原子级分散rh催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散rh催化剂均在139℃下催化co完全转化。
71.应用例4
72.基本同应用例1,分别将50mg实施例4制备的碳包覆的氧化铈负载原子级分散ir催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散ir催化剂与500mg石英砂
混合装入固定床反应器,都以15000ml
·
g-1
·
h-1
的空速通入比例为1vol.%co+20vol.%o2+n2的气体,根据升温程序升高温度进行反应。碳包覆的氧化铈负载原子级分散ir催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散ir催化剂均在147℃下催化co完全转化。
73.应用例5
74.基本同应用例1,分别将50mg实施例5制备的碳包覆的氧化铈负载原子级分散ru催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散ru催化剂与500mg石英砂混合装入固定床反应器,都以15000ml
·
g-1
·
h-1
的空速通入比例为1vol.%co+20vol.%o2+n2的气体,根据升温程序升高温度进行反应。碳包覆的氧化铈负载原子级分散ru催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散ru催化剂均在150℃下催化co完全转化。
75.应用例6
76.基本同应用例1,分别将50mg实施例6制备的碳包覆的氧化铈负载原子级分散pt催化剂和经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂与500mg石英砂混合装入固定床反应器,都以15000ml
·
g-1
·
h-1
的空速通入比例为1vol.%co+20vol.%o2+n2的气体,根据升温程序升高温度进行反应。碳包覆的氧化铈负载原子级分散pt催化剂在、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂均在110℃下催化co完全转化。
77.应用例7
78.基本同应用例1,分别将50mg实施例7制备的碳包覆的氧化铈负载原子级分散pt催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂与500mg石英砂混合装入固定床反应器,都以15000ml
·
g-1
·
h-1
的空速通入比例为1vol.%co+20vol.%o2+n2的气体,根据升温程序升高温度进行反应。碳包覆的氧化铈负载原子级分散pt催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂均在115℃下催化co完全转化。
79.应用例8
80.基本同应用例1,分别将50mg实施例8制备的碳包覆的氧化铈负载原子级分散pt催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂与500mg石英砂混合装入固定床反应器,都以15000ml
·
g-1
·
h-1
的空速通入比例为1vol.%co+20vol.%o2+n2的气体,根据升温程序升高温度进行反应。碳包覆的氧化铈负载原子级分散pt催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂均在130℃下催化co完全转化。
81.应用例9
82.基本同应用例1,分别将50mg实施例9制备的碳包覆的氧化铈负载原子级分散pt催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂与500mg石英砂混合装入固定床反应器,都以15000ml
·
g-1
·
h-1
的空速通入比例为1vol.%co+20vol.%o2+n2的气体,根据升温程序升高温度进行反应。碳包覆的氧化铈负载原子级分散pt催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂均在128℃下催化co完全转化。
83.应用例10
84.基本同应用例1,分别将50mg实施例10制备的碳包覆的氧化铈负载原子级分散pt催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂与500mg石英砂混合装入固定床反应器,都以15000ml
·
g-1
·
h-1
的空速通入比例为1vol.%co+20vol.%o2+n2的气体,根据升温程序升高温度进行反应。碳包覆的氧化铈负载原子级分散pt催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂均在160℃下催化co完全转化。
85.图4为本发明实施例1、8-10制备的(pt1/ceo2)@c、pt1/ceo2和ceo2催化co氧化的反应性能图。从图4可以看出,pt1/ceo2和儿茶酚胺的质量比为5:1时,(pt1/ceo2)@c-5:1在170℃催化co完全转化;pt1/ceo2和儿茶酚胺的质量比为40:1时,(pt1/ceo2)@c-40:1在130℃催化co完全转化;pt1/ceo2和儿茶酚胺的质量比为10:1时,(pt1/ceo2)@c-10:1在130℃催化co完全转化;pt1/ceo2和儿茶酚胺的质量比为20:1时,(pt1/ceo2)@c-20:1在100℃催化co完全转化。通过调节pt1/ceo2和儿茶酚胺的质量比,可以增强催化剂的低温活性。
86.应用例11
87.基本同应用例1,分别将50mg实施例6制备的碳包覆的氧化铈负载原子级分散pt催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂与500mg石英砂混合装入固定床反应器,都以18000ml
·
g-1
·
h-1
的空速通入比例为1vol.%co+20vol.%o2+n2的气体,根据升温程序升高温度进行反应。碳包覆的氧化铈负载原子级分散pt催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂均在118℃下催化co完全转化。
88.应用例12
89.基本同应用例1,分别将50mg实施例7制备的碳包覆的氧化铈负载原子级分散pt催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂与500mg石英砂混合装入固定床反应器,都以18000ml
·
g-1
·
h-1
的空速通入比例为1vol.%co+20vol.%o2+n2的气体,根据升温程序升高温度进行反应。碳包覆的氧化铈负载原子级分散pt催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂均在125℃下催化co完全转化。
90.应用例13
91.基本同应用例1,分别将50mg实施例8制备的碳包覆的氧化铈负载原子级分散pt催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂与500mg石英砂混合装入固定床反应器,都以18000ml
·
g-1
·
h-1
的空速通入比例为1vol.%co+20vol.%o2+n2的气体,根据升温程序升高温度进行反应。碳包覆的氧化铈负载原子级分散pt催化剂、经400℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂均在140℃下催化co完全转化。
92.应用例14
93.基本同应用例1,分别将50mg实施例11制备的碳包覆的氧化铈负载原子级分散pt催化剂、经300℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂与500mg石英砂混合装入固定床反应器,都以15000ml
·
g-1
·
h-1
的空速通入比例为1vol.%co+20vol.%o2+n2的气体,根据升温程序升高温度进行反应。碳包覆的氧化铈负载原子级分散pt催化剂、
经300℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂均在100℃下催化co完全转化。
94.应用例15
95.基本同应用例1,分别将50mg实施例12制备的碳包覆的氧化铈负载原子级分散pt催化剂、经500℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂与500mg石英砂混合装入固定床反应器,都以15000ml
·
g-1
·
h-1
的空速通入比例为1vol.%co+20vol.%o2+n2的气体,根据升温程序升高温度进行反应。碳包覆的氧化铈负载原子级分散pt催化剂、经500℃下h2热还原处理的碳包覆的氧化铈负载原子级分散pt催化剂均在100℃下催化co完全转化。
96.对比例
97.将ceo2分散在100ml超纯水(18.2mω)中,超声振动使ceo2均匀分散,调节分散液ph为8.5,加入质量为ceo2质量1/20的多巴胺,搅拌2h后离心混合溶液并洗涤得到的固体,在60℃下干燥12h,之后,在ar气氛下以5℃/min的升温速率升至600℃保持2h,得到具有包覆层的氧化铈载体(ceo2)@c。
98.对比应用例
99.基本同应用例1,将50mg对比例制备的具有包覆层的氧化铈载体(ceo2)@c与500mg石英砂混合装入固定床反应器,以15000ml
·
g-1
·
h-1
的空速通入比例为1vol.%co+20vol.%o2+n2的气体,根据升温程序升高温度进行反应。
100.图5为实施例1的(pt1/ceo2)@c、pt1/ceo2、ceo2和对比例的(ceo2)@c催化co氧化的反应性能图。从图5可以看出,具有包覆层的氧化铈载体(ceo2)@c在350℃下催化co完全转化,而碳包覆的氧化铈负载原子级分散pt催化剂在100℃下催化co完全转化,说明原子级分散金属中心配位结构pt
1-o
2-n2是低温下高活性的催化中心,仅包覆层与ceo2载体无法构成低温下高活性的催化中心。
101.显然,上述实施例仅仅是为清楚地说明所作的举例,并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1