一种提高一步法制备六硝基芪得率的方法
1.本发明属于含能材料技术领域,涉及一种用电化学法合成六硝基芪的方法。
背景技术:
2.六硝基芪(hns)作为一种具低蒸汽压、低敏感性和较高热安定性的耐热炸药,被广泛应用于石油勘探、太空和深海研究。目前,生产hns的方法主要有一步法和两步法两种,其中两步法由于其较高的产率和产品纯度,成为国内主要的生产hns的方法。相比于两步法,一步法主要的缺陷是产率低,副产物较多,但是,一步法反应操作简单,易于连续化,这非常有利于hns的大规模连续化生产。
3.根据先前的报道,shipp等向冷却至0℃的的四氢呋喃/甲醇溶液中快速加入次氯酸钠溶液并保持反应温度为,然后用丙酮冲洗得粗品,该方法制备产率比较低,通常只有30
‑
35%(j.org.chem.1964,29,2620
‑
2623)。salter等在对上述方法进行了改进,他们在反应液中加入含氮碱,其推荐的是有机胺的盐酸盐,使hns的得率提高到45%左右,但是含氮碱的使用带来了后处理的不便(def sci.,vol 31,1980,4(10):305
‑
308)。golding等对上述方法进行了改进,即在反应中加入钙的氧化物,能减少副反应。他们称hns的得率可达46%,但此法反应时间达到几十个小时且需加酸处理才能得到粗品(us patent:5023386,1991
‑
07
‑
11)。
4.尽管有很多文献对应用一步法制备六硝基芪进行过报道,但大多数这些方法都存在以下一个或多个缺点:反应时间长,产物收率低,反应成本高和副反应多、后处理困难等。同时,六硝基芪在合成过程中的强放热使得体系的温度变化较大,对产物的产率、实验的安全性均有较大影响,这个问题一直没有得到解决。
技术实现要素:
5.本发明的目的在于提供一种提高一步法制备六硝基芪得率的方法。
6.为达到上述目的,本发明的技术方案是:一种用电化学法合成六硝基芪的方法,包括的具体步骤:
7.(1)在四氢呋喃和甲醇混合溶剂存在下,将三硝基甲苯(tnt)与次氯酸钠通过静态混合器混合,收集混合后的反应液反应一段时间,
[0008][0009]
(2)将步骤(1)反应后溶液加入溶剂、电解质、脱氢剂,继续电催化反应一段时间,得到目标产物的步骤,
[0010][0011]
进一步的,步骤(1)中,将三硝基甲苯(tnt)与次氯酸钠预冷至0℃后通过静态混合器混合,收集混合后的反应液反应1~1.5小时。
[0012]
进一步的,步骤(1)中,四氢呋喃和甲醇的体积比为2:1。
[0013]
进一步的,步骤(1)中,以摩尔比计,次氯酸钠:三硝基甲苯控制在1:1到1.5:1之间,次氯酸钠中的有效氯含量在2%~8%之间。
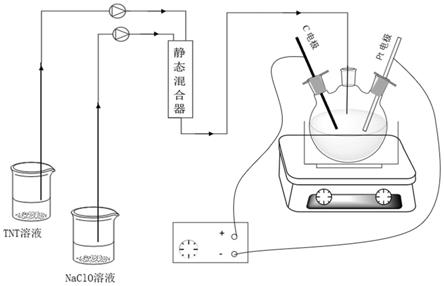
[0014]
进一步的,步骤(1)中,反应液的ph控制在10
‑
10.5之间,反应液的温度控制10
‑
20℃。
[0015]
进一步的,步骤(2)中,溶剂选自乙醚、dmso、丙酮、苯、1,4
‑
二氧六环中的任意一种。
[0016]
进一步的,步骤(2)中,脱氢剂选自三乙烯二胺(dabco)、三乙胺、fecl2‑
tempo、dipea等任意一种,其添加量与步骤(1)反应后溶液中副产物hnbb的摩尔量相同。
[0017]
进一步的,步骤(2)中,电解质选自四甲基四氟硼酸铵、四丁基高氯酸铵、四丁基四氟硼酸胺、高氯酸铵和四丁基六氟磷酸胺中任意一种。
[0018]
进一步的,步骤(2)中,电催化反应以石墨棒为阳极,铂片为阴极,在恒定电流4
‑
12ma下通电进行。
[0019]
进一步的,步骤(2)中,电催化反应温度为10
‑
20℃,反应时间为0.5~1小时。
[0020]
与现有技术相比,本发明的优点是:本发明采用了静态混合器,有效的克服了在合成过程中的强放热使得体系温度变化较大的问题,使得反应的产率和安全性均得到改善;另外,采用电催化模式,反应过程简单,安全环保,生成的六硝基芪产率高,有望成为一种新型的制备hns的方法。
附图说明
[0021]
图1含静态混合器的电化学催化系统装置图。
[0022]
图2为该反应机理推测图。
[0023]
图3为反应产物精制后的液相分析图。
[0024]
图4为反应产物的核磁共振1h图。
[0025]
图5为反应产物精制后的核磁共振1h图。
[0026]
图6为反应产物的核磁共振
13
c图。
具体实施方式
[0027]
下面通过附图和实施例对本发明的技术方案做出进一步的具体说明。
[0028]
结合图1,本发明反应体系的装置包括含静态混合器和电化学反应系统。反应物料通过两台蠕动泵经过聚四氟乙烯管输送至混合三通。混合三通直接连接到静态混合器的顶部,从静态混合器底部流出的物料由聚四氟乙烯管输送至后续电化学反应系统。物料在进入静态混合器之前需先进行预冷。物料在静态混合器中混合的时间不超过3分钟。
[0029]
本发明所述的一步法制备hns的方法,首先将原料送入静态混合器进行混合,混合
液反应1.5小时后得到大部分产物,然后将反应后溶液通过电催化的方法,使得副产物六硝基联苄(hnbb)脱氢转化为产物六硝基芪(hns),具体步骤是:从静态混合器出来的混合液即反应液用三口烧瓶收集,在10~20℃的温度下,并通过ph计调节体系ph,使得体系ph维持在10
‑
10.5(通过加入25wt%h2so4和6wt%naoh调节),反应液先反应1~1.5小时。然后用电催化的方法将反应后的副产物进一步转化为目标产物,具体过程是:加入电解质,用dmso溶解副产物中的hnbb,实验温度保持20℃左右,施加一定电流,以石墨棒为阳极,铂片为阴极,并用三乙烯二胺(dabco)作为脱氢剂,反应搅拌0.5~1小时。反应结束后,混合液过滤并用水洗涤三次后置于60℃烘箱干燥,得到hns粗品。粗品经丙酮煮解,重结晶后抽滤,用甲醇、水洗涤后得到黄色粉末状固体。通过液相分析和熔点测量等手段确定产物的纯度。
[0030]
本发明采用的静态混合器为sk静态混合器,由启东市长江化工机械有限公司提供。
[0031]
实施例1:单独应用静态混合器对一步法合成六硝基芪进行优化
[0032]
首先取用一个带搅拌子的烘箱干燥后的三口烧瓶(150ml)中,三口烧瓶置于水浴锅中,易于进行温度调节。称取2.0g的三硝基甲苯(tnt),溶解30ml四氢呋喃(thf)和15ml甲醇(meoh)中,通过蠕动泵泵入混合三通。量取16ml次氯酸钠加水稀释至50ml,通过蠕动泵泵入混合三通。两股反应物料配制好后置于冰水中预冷,预冷完成后打开混合三通的入口阀,按照预定的流速设置蠕动泵的参数,同时接通两个泵。将离开出口管的混合液收集在准备好的三口烧瓶(磁力搅拌)中,当最后的反应液离开混合器时,关闭两个泵,此时停止收集混合液。然后对混合器系统进行冲洗。先将出口管移到废液收集装置,然后开启泵,通过tnt溶液入口系统用20ml甲醇溶液、30ml水冲洗系统,通过naclo入口系统用50ml冲洗系统,待清洗液流干后关闭泵。混合液反应2.5小时后,使用布氏漏斗减压抽滤,然后用去离子水清洗三次后置于60℃烘箱干燥,得到粗品(产率为38%,纯度也较低)。粗品经丙酮煮解,重结晶进行精制。通过液相分析和熔点测量等手段确定产物的纯度。精制后产物的核磁表征(见附图4)为:1h nmr(500mhz,dmso
‑
d6)δ=3.42
‑
3.39(s,4h,ch2),7.15
‑
7.13(s,2h),9.10
‑
9.04(s,4h,aryl h),9.20
‑
9.11(s,4h)。
[0033]
下面为了提高该过程的产率,这里以反应温度、反应体系ph、反应物的摩尔比(n(naclo):n(tnt))、次氯酸钠的有效氯为正交试验的因素,采用lg(34)正交表来进行试验,结果如下:
[0034]
表1影响产率的正交水平因素表
[0035][0036]
表2影响产率的正交水平实验表
[0037]
[0038][0039]
从上表可以看出,因素a、b、c、d的极差分别为3、5.2、5.7、4.2,可知反应体系的ph对hns产率的影响最大;另外,可以知道单独应用静态混合器一步法合成六硝基芪过程的最佳反应条件为a2b2c3d2,根据正交实验结果进行验证实验,通过控制适当的n(naclo):n(tnt)、次氯酸钠有效氯含量、反应温度、反应体系ph。可以得到单独应用静态混合器一步法合成六硝基芪过程的最佳反应条件:溶剂投料比为四氢呋喃:甲醇=2:1,tnt与次氯酸钠溶液的投料摩尔比为n(tnt):n(naclo)=1:1.2(tnt称取2g,次氯酸钠溶液量取16ml);反应温度保持在15℃左右,反应持续时间以2~3小时为宜;次氯酸钠有效氯含量为5%;反应开始后使用25wt%h2so4和6wt%naoh调节体系ph使之保持在10.5左右。在这些条件之下,反应的粗产率为47%。对粗品对粗产物进行液相分析,发现含有相当含量的hnbb以副产物的形式存在。然后对粗品进行精制,再通过测定熔点(308.4℃)与核磁共振分析,确认其为目标产物2,2',4,4',6,6'
‑
六硝基二苯基乙烯(hns)。
[0040]
实施例2:单独应用电催化的方法对一步法合成六硝基芪过程进行优化
[0041]
首先称取2.0gtnt(约8.8mmol),将tnt溶解于事先量好的15ml甲醇与30ml四氢呋喃中。配置好的tnt溶液置于冰水浴中降温至0~5℃。量取16ml有效氯含量为5%的次氯酸钠溶液,加水稀释至50ml,倒入固定在低温冷却装置上的圆底烧瓶(150ml)中,预冷至0~5℃。在适当的搅拌转速下,将烧杯内的tnt溶液迅速倒入烧瓶中。烧瓶内溶液颜色由淡黄快速转变为紫红色,温度剂读数很快上升至25℃左右,当温度回落稳定时撤去低温反应装置,并保证适当的反应温度大约1~2h。持续搅拌后,可以明显看出烧瓶中有固体析出。此时加入n
‑
bu4nbf4等电解质,用dmso等溶剂溶解副产物中的hnbb(hnbb要溶解在溶液中才能进行电解反应),实验温度保持25℃左右。在三口烧瓶上装石墨棒(φ=6mm)作为阳极,铂片电极(10mm
×
10mm)作为阴极。并用三乙烯二胺(dabco)作为脱氢剂。将反应混合物在4~12ma的恒定电流下电解0.5~1小时。反应结束后,混合液使用布氏漏斗减压抽滤,然后用去离子水清洗三次后置于60℃烘箱干燥,得到粗品(产率为37.2%)。粗品经丙酮煮解,重结晶进行精制。通过液相分析和熔点(285.4℃)测量确定产物的纯度。
[0042]
下面为了提高该过程的产率,分别从电解质种类、溶剂种类、脱氢剂种类、反应体系的恒定电流/电压等角度对该反应过程进行了优化。
[0043]
(1)反应具体步骤如上所述,反应混合液反应1~1.5h后,进行电化学催化,选用适当的溶剂(溶解hnbb)、脱氢剂,反应电流/电压恒定在适当的范围,在反应体系中加入不同的电解质,并对每次实验的产物进行分析,电解质种类对反应产率的影响如表3所示。由表3可以清晰的发现,若电催化过程中未加入电解质,所得产物的纯度和纯度均较低。加入的电解质中,大部分是季铵盐,主要是因为季铵盐作为常用的有机支持电解质,具有导电性能好、化学性质稳定、价格低廉、易于制取等优点。在其他条件不做改变的情况下,可以发现,在电解质的作用下,反应的产率都有了提高,主要是因为副产物中的hnbb转化为目标产物hns。
[0044]
表3不同种类的电解质对产率的影响
[0045][0046]
注:纯品hns的熔点为312.4℃,测量所得产物的熔点可以大概反应其纯度,当然必须通过液相分析等手段才能得到产物准确的纯度。
[0047]
(2)反应具体步骤如上所述,反应混合液反应1~1.5h后,进行电化学催化,选用适当的电解质、脱氢剂,反应电流/电压恒定在适当的范围,在反应体系中加入不同的溶剂,并对每次实验的产物进行分析,溶剂对反应产率的影响如表4所示。可以发现,用dmso作为溶剂时,反应的产率较高,这主要是由于hnbb在dmso中溶解彻底(dmso的介电常数较大,极性较大),这使得电解反应更易进行。一个很明显的对比是,虽然hnbb也溶于1,4
‑
二氧六环,但是其介电常数较小,这使得hnbb难以参加电解反应。另外,丙酮的沸点太低,不适合做溶剂。而当使用酰胺(dmf,六甲基磷酰胺)为反应溶剂时,液相检测产品中有大量不确定的副产物。
[0048]
表4不同种类的溶剂对产率的影响
[0049][0050]
(3)反应具体步骤如上所述,反应混合液反应1~1.5h后,进行电化学催化,选用适当的电解质、溶剂,反应电流/电压恒定在适当的范围,在反应体系中加入不同的脱氢剂,并对每次实验的产物进行分析,溶剂对反应产率的影响如表5所示。可以发现,加入的脱氢剂,大部分是叔胺,主要是因为叔胺作为脱氢剂,具有化学性质稳定、价格低廉、易于制取、脱氢效果好(叔胺容易与hnbb上苯环与亚甲基相连的碳原子结合,使得亚甲基上的氢原子脱去而形成二价阴离子hns)等优点。在其他条件不做改变的情况下,可以发现,在脱氢剂的作用下,反应的产率都有了提高,主要是因为副产物中的hnbb转化为目标产物hns。其中dabco(三乙烯二胺)作为电解质时,产物产率较高,且通过测量熔点与液相分析可以发现产品的纯度也较高。这主要是因为一个dabco中含有两个三级n原子,相同量的脱氢剂,dabco更容易使hnbb脱去两个质子,形成中间体hns2‑
(见附图),另外,dabco的碱强度中等(pk
a
≈9),对反应体系的ph影响不大。fecl2‑
tempo作为脱氢剂需要在氧气的氛围中才能进行,且对反应体系的温度也有要求,不适合做脱氢剂。
[0051]
表5不同种类的脱氢剂对产率的影响
[0052][0053]
(4)反应具体步骤如上所述,反应混合液反应1~1.5h后,进行电化学催化,选用适当的电解质、溶剂、脱氢剂,保持反应体系电流/电压恒定在适当的范围,并对每次实验的产物进行分析,通过表6可以发现,当反应体系的恒定电流低于2ma高于12ma时,反应收率迅速下降。通过上图还可以发现,反应体系的恒定电流在6~10ma之间时,产物中hns产率较高,通过液相分析亦可发现该范围内hns的纯度亦较高。
[0054]
表6反应体系的恒定电流对反应产率的影响
[0055][0056]
(4)选用适当的电解质、脱氢剂、溶剂,反应电流/电压恒定在适当的范围,可以得到单独应用电催化的方法对一步法合成六硝基芪过程的最佳工艺条件:以四丁基高氯酸铵作为电解质,用dmso等溶剂溶解副产物中的hnbb,实验温度保持25℃左右。在三口烧瓶上装石墨棒(φ=6mm)作为阳极,铂片电极(10mm
×
10mm)作为阴极。并用三乙烯二胺(dabco)作为脱氢剂,将反应混合物在8ma的恒定电流下电解0.5
‑
1小时。在这些条件之下,反应的粗产率为48.5%。对粗品对粗产物进行液相分析,发现产品纯度较高。然后对粗品进行精制,再通过测定熔点(310.4℃)与核磁共振分析,确认其为目标产物2,2',4,4',6,6'
‑
六硝基二苯基乙烯(hns)。
[0057]
实施例3
[0058]
综合应用静态混合器和电化学系统对一步法合成六硝基芪进行优化
[0059]
根据实施例1、2得到的结果,综合应用静态混合器和电化学系统对一步法合成六硝基芪进行优化,具体过程为:两股物料通过蠕动泵进入静态混合器,混合时间<3min,用三口烧瓶收集静态混合器出口管的混合液,混合液反应1~1.5h在反应前1~1.5h,溶剂投料比为四氢呋喃:甲醇=2:1,tnt与次氯酸钠溶液的投料摩尔比为n(tnt):n(naclo)=1:1.2(tnt称取2g,次氯酸钠溶液量取16ml);反应温度保持在15℃左右;次氯酸钠有效氯含量为5%;反应开始后使用25wt%h2so4和6wt%naoh调节体系ph使之保持在10~10.5之间。在反应后0.5~1h,以四丁基高氯酸铵作为电解质,用dmso等溶剂溶解副产物中的hnbb,实验温度保持20℃左右。在三口烧瓶上装石墨棒(φ=6mm)作为阳极,铂片电极(10mm
×
10mm)作为阴极。并用三乙烯二胺(dabco)作为脱氢剂,将反应混合物在9ma的恒定电流下电解0.5~1小时。反应结束后,用布氏漏斗减压抽滤,然后用去离子水清洗三次后置于60℃烘箱干燥,得到粗品。粗品经丙酮煮解,重结晶进行精制。通过液相分析和熔点测量等手段确定产物的纯度。在这些条件之下,反应的产率为53%。对精制后的产物进行液相分析(见附图3),发现产品纯度较高。然再通过测定熔点(312.8℃)与核磁共振分析(见附图5、6),1h nmr(500mhz,dmso
‑
d6)δ=7.40
‑
7.35(s,2h),9.20
‑
9.15(s,4h)。确认其为目标产物2,2',4,4',6,6'
‑
六硝基二苯基乙烯。
[0060]
本发明基于实验的结果,提出了hnbb电催化转化为hns的反应机制,具体反应历程见附图2。
[0061][0062]
由于hnbb上六个硝基基团产生的影响,胺首先结合到苯环上与亚甲基相连的碳原子上,hns二价阴离子产生主要是因为以下原因:1)亚硝酰基的吸电子能力,它可以稳定负电荷;(2)通过形成二价阴离子可以缓解苯环上众多硝基的空间拥挤。另外,二价阴离子的结构允许围绕中心碳
‑
碳键自由旋转,使得hns二价阴离子(hns2‑
)能相对稳定的存在。然后,在恒定电流/电压的作用下,hns2‑
在石墨(anode)电极上电解生成hns。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1