一种以聚吡咯凝胶电解质作为支持电解质的芳香醇电化学转化方法
1.本发明属于有机电合成领域,本发明涉及一种以聚吡咯凝胶电解质作为支持电解质的芳香醇电化学转化方法。
背景技术:
2.在有机电化学合成中,大部分有机溶剂的电阻很高,不仅增大了电能的消耗,而且会导致反应体系温度升高,引起副反应的发生。为此往往需要加入过量的支持电解质,以降低反应体系的电阻。常用的支持电解质有liclo4、naclo4、r4n
+
clo
4-、r4n
+
x-等作为支持电解质,但这类传统支持电解质不仅价格昂贵,而且其加入量非常大,常常是反应底物的数倍;此外,支持电解质后处理困难,难以回收利用,易造成相应的浪费以及环境污染。因此,开发易回收利用的电解质来减少传统支持电解质的使用量是一个必然趋势。
3.针对传统支持电解质在电合成应用中的短板,yoshida等(the journal of organic chemistry,1980,45:5269-5273)使用珠状树脂——由聚4-乙烯基吡啶氢溴酸盐在电化学作用下生成的聚合物试剂进行仲醇的氧化。该珠状树脂作为支持电解质可以回收再利用。francke等(angewandte chemie-international edition,2018,57(2):422-426)开发了一种可溶的聚(甲基丙烯酸酯)电解质,将其用于醇的电氧化,并且通过透析或者超滤即可回收重复利用。但这些聚电解质存在制备工艺复杂、时间长等问题。
4.在聚吡咯、聚苯胺等导电聚合物可被用于太阳能电池中,来降低反应体系的电荷转移电阻,提高离子电导率以及对i-/i
3-氧化还原电对的电催化活性(journal of power sources 2014,254,98-105)。聚吡咯凝胶制备工艺简单、易回收利用,但其作为电解质在有机化合物的电化学转化中的应用未见报道,同时其微孔结构对不同有机反应底物的适应性有待于进一步提高。本发明采用掺杂方法调整聚吡咯凝胶结构,改善有机电合成反应体系中高电阻的问题,并将其应用于芳香醇电化学转化体系中,既可以减少传统支持电解质的使用量,又进一步提高芳香醇的电氧化转化率。
技术实现要素:
5.本发明提供一种以聚吡咯凝胶电解质作为支持电解质的芳香醇电化学转化方法,在减少传统支持电解质用量的同时,有效提高芳香醇的电氧化转化率及生成相应芳香醛的选择性。
6.为解决上述技术问题,本发明采用如下技术方案:
7.一种以聚吡咯凝胶电解质作为支持电解质的式(ⅰ)所示的芳香醇电化学转化方法,依次包括如下步骤:
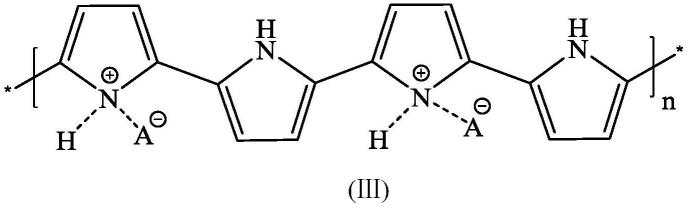
8.步骤一:将含有芳香醇、聚吡咯凝胶电解质、传统支持电解质和有机溶剂混合作为电解液,所述电解液中,芳香醇、聚吡咯凝胶电解质、传统支持电解质的初始浓度分别为1~13.8g/l、0.5~2g/l、0.005~0.05mol/l,将电解液加入无隔膜电解槽中;所述的聚吡咯凝
胶无掺杂或者是十二烷基苯磺酸钠(dbsna)、十二烷基苯磺酸(dbsa)或柠檬黄(tz)掺杂的聚吡咯凝胶,其典型结构如式(iii)所示;
[0009][0010]
式(ⅲ)中,a为掺杂剂或h,所述的掺杂剂为十二烷基苯磺酸钠(dbsna)、十二烷基苯磺酸(dbsa)或柠檬黄(tz)等;
[0011]
步骤二:将阴极材料和阳极材料放入无隔膜电解槽中;
[0012]
步骤三:将阴极材料和阳极材料连接稳压直流电源进行芳香醇电化学转化反应,生成式(ⅱ)所示的芳香醛;
[0013]
反应方程式如下:
[0014][0015]
式(ⅰ)或(ⅱ)中,r为c1-c4的烷基、c1-c4的烷氧基、卤素取代的c1-c4的烷基或卤素。
[0016]
作为优选,r为och3、ch3、t-bu、cl或cf3。
[0017]
作为优选,所述有机溶液为乙腈和n,n二甲基甲酰胺中的一种,更优选为n,n二甲基甲酰胺。
[0018]
作为优选,所述式(ⅰ)所示的聚吡咯凝胶电解质中,a为掺杂剂,更优选a为dbsa。
[0019]
作为优选,所述电解液中,聚吡咯凝胶电解质的初始浓度为0.8~1.5g/l,更优选为1g/l。
[0020]
作为优选,所述传统支持电解质为高氯酸锂(liclo4)和四丁基高氯酸铵(tbap)中的一种,更优选为liclo4。
[0021]
作为优选,所述电解液中,所述传统支持电解质的初始浓度为0.005~0.01mol/l。
[0022]
作为优选,所述的无隔膜电解槽为带夹套可加热的单室电解槽。
[0023]
作为优选,所述阳极材料为碳棒或铂片,优选为铂片。
[0024]
作为优选,所以阴极材料为碳棒或铅片,优选为铅片。
[0025]
作为优选,所述芳香醇电化学转化反应过程中,电流大小为5~40ma,更优选为5~20ma,最优选为10ma。
[0026]
作为优选,所述芳香醇电化学转化反应温度控制为30℃~70℃,更优选为50~70℃,最优选60℃。
[0027]
作为优选,所述芳香醇电氧化转化反应搅拌下进行,搅拌速率为100rpm~800rpm,更优选为400~600rpm,最优选500rpm。
[0028]
作为优选,所述芳香醇电化学转化反应在搅拌下进行,电流大小控制为5~40ma,
反应温度控制为30℃~70℃,搅拌速率为100rpm~800rpm,反应时间为3-8h。
[0029]
本发明特别优选所述芳香醇电化学转化反应在搅拌下进行,电流大小控制为10ma,反应温度控制为60℃,搅拌速度为500rpm,反应时间为3~8h。
[0030]
本发明所述的聚吡咯凝胶可参照文献报道的方法进行制备。通常制备步骤如下所示:配制含有掺杂剂、吡咯单体和溶剂(如异丙醇、乙腈等)的混合溶液,快速倒入氧化剂(如(nh4)2s2o8、fecl3、h2o2等)溶液引发聚合反应,充分聚合后;将聚吡咯水凝胶依次浸入乙醇(如纯化12h)和去离子水(如纯化12h)中纯化,再放入烘箱干燥后,最后经研磨后制得聚吡咯凝胶电解质。其中,掺杂剂与吡咯单体的摩尔比一般为0.02~0.17:1。
[0031]
本发明的有益效果为:
[0032]
(1)聚吡咯凝胶电解质制备工艺要求较低、后处理简单,通过过滤即可实现回收利用,减少资源的浪费,节约成本。
[0033]
(2)聚吡咯凝胶与传统支持电解质之间的“协同作用”极大降低了有机电化学反应对传统支持电解质的依赖。聚吡咯凝胶电解质与极少量的传统支持电解质就可以使电化学反应快速高效地进行,且反应具有很好的选择性。
[0034]
(3)该方法收率高,原子经济性强,不产生有害物质,对环境友好,极大地减少了传统支持电解质的使用量,减少了资源的浪费,降低了生产成本。
附图说明
[0035]
图1为p-meobzoh在不同支持电解质中的循环伏安图。
具体实施方式
[0036]
为了更好地理解本发明,以下是本发明的具体实施例结合相关附图,对本发明的技术方案作进一步的描述,但本发明并不仅限于这些实施例。
[0037]
实施例中所使用的聚吡咯凝胶电解质依据如下文献中的方法进行制备:
[0038]
无掺杂的聚吡咯凝胶:nano letters,2015,15(11),7736
–
7741;
[0039]
聚吡咯凝胶(tz):journal of electroanalytical chemistry.2019,832,174-181;
[0040]
聚吡咯凝胶(dbsa、nadbs):polym eng sci.2013;53(11):2465-2469。
[0041]
下述实施例所用的芳香醇的结构式如式(1-1)~(1-5)所示:
[0042][0043]
对应目标产物芳香醛的结构如式(2-1)~(2-5)所示:
[0044][0045]
实施例0:
[0046]
为了研究聚吡咯凝胶电解质对反应是否有促进作用,能否替代传统支持电解,首先进行了p-meobzoh在不同支持电解质中的循环伏安研究,测试结果如图1所示。该循环伏安测试在标准三电极体系中进行,以铂盘电极为工作电极,铂片(20
×
20
×
0.2mm)为对电极,以ag/agno3(0.01m agno3)为参比电极。扫描速率为50mv/s,测试温度为25℃,溶剂乙腈10ml,p-meobzoh添加量为0.5mmol。只有聚吡咯凝胶(dbsa)作为支持电解质时,反应体系中没有出现氧化还原峰。当加入50mmol/l liclo4作为支持电解质后,体系中出现了相应的氧化峰。当向反应体系中同时加入50mmol/l liclo4和0.01g聚吡咯凝胶电解质后,相应氧化峰明显增高,且峰电位也有所负移,表明聚吡咯凝胶电解质会吸附溶液中liclo4形成复合电解质,复合电解质与溶液中剩余的liclo4进一步产生“协同作用”,能够加快反应速率。因此,聚吡咯凝胶电解质在一定程度上可部分替代传统支持电解质。
[0047]
以芳香醇为原料制备芳香醛的电解步骤与结果如下:
[0048]
实施例1:
[0049]
在50ml无隔膜电解槽中加入0.069g p-meobzoh,0.0318g(10mmol/l)liclo4,30ml溶剂(n,n二甲基甲酰胺),反应温度60℃,以pt作阳极,以pb作阴极,再加入6mm
×
10mm尺寸搅拌子,磁力搅拌设置为500rpm,启动直流电源控制电流为10ma,电解4h,得到目标产物对甲氧基苯甲醛。电解产物的收率通过气相色谱gc进行分析,分析方法为面积归一法,产物收率如表1所示为69.5%。实施例2-5、对比例1:
[0050]
反应步骤同实施例1,所不同的是额外将不同的聚吡咯凝胶电解质加入到电解液中,分别是无掺杂剂的聚吡咯凝胶电解质0.03g(实施例2)、dbsna为掺杂剂的聚吡咯凝胶电解质0.03g(实施例3)、dbsa为掺杂剂的聚吡咯凝胶电解质0.03g(实施例4)、tz为掺杂剂的聚吡咯凝胶电解质0.03g(实施例5)以及liclo4加入量为0.477g(150mmol/l)(对比例1),进行上述恒电流电解实验,结果列于表1中。
[0051]
表1不同支持电解质作用下p-meobzoh电氧化生成对甲氧基苯甲醛情况
[0052][0053]
由表1可知,在高浓度liclo4下(对比例1),不仅反应底物的转化率下降,而且目标产物的收率只有43.3%。而在较低浓度的liclo4(实施例1)时反应底物的转化率和目标产物收率均有所提高。但加入了聚吡咯凝胶电解质后,反应的目标产物收率明显提高。并且聚吡咯凝胶电解质有无掺杂,目标产物的收率都高达95%以上。因此,聚吡咯凝胶电解质的加入有效提高了传统支持电解质的作用,促进了芳香醇电化学转化反应的顺利进行。
[0054]
实施例6-15、对比例6:
[0055]
反应步骤同实施例1,所不同的是将所有电解实验中liclo4的使用量降低至0.0159g(5mmol/l)或不添加liclo4以及不通电的情况(对比例6),相应的聚吡咯凝胶电解质的添加量仍为0.03g,进行上述恒电流电解实验,结果列于表2。
[0056]
表2不同liclo4浓度下p-meobzoh生成对甲氧基苯甲醛
[0057][0058]
由上述反应结果可知,只添加聚吡咯凝胶电解质作为支持电解质时(实施例7,9,11,13,15),可认为没有发生相应的电化学转化,而只进行p-meobzoh与溶解氧的氧化反应;在极低浓度的liclo4下,加入聚吡咯凝胶后,大部分实施例表现出电解液的电阻偏大,无法使电化学反应有效进行(实施例8,10,14);而在dbsa掺杂的聚吡咯凝胶电解质作用下,只需要5mmol/l liclo4即可使目标产物的收率提高至97.6%,因此聚吡咯凝胶电解质优选dbsa为掺杂剂。
[0059]
实施例16:
[0060]
在50ml无隔膜电解槽中加入0.069g p-meobzoh,0.0159g(5mmol/l)liclo4,0.03gdbsa掺杂的聚吡咯凝胶电解质,30ml溶剂(n,n二甲基甲酰胺),反应温度60℃,以碳棒
作阳极,以碳棒作阴极,再加入6mm
×
10mm尺寸搅拌子,磁力搅拌设置为500rpm,启动直流电源控制电流为10ma,电解4h,得到目标产物对甲氧基苯甲醛。电解产物收率通过气相色谱gc进行分析,分析方法为面积归一法,产物收率如表3所示为83.3%。
[0061]
实施例17-19:
[0062]
反应步骤同实施例16,所不同的是聚吡咯凝胶电解质掺杂剂分别为无掺杂(实施例17)、dbsna(实施例18)、tz(实施例19),进行上述恒电流电解实验,结果列于表3。
[0063]
表3不同聚吡咯凝胶电解质作用下p-meobzoh合成对甲氧基苯甲醛
[0064][0065]
由上述反应结果可知,在低浓度的liclo4为支持电解质时,加入有掺杂剂的聚吡咯凝胶电解质后,以碳棒作为阴极和阳极也有较好的效果,目标产物的收率≥76.2%;相较于pt/pb电极对,dbsa掺杂的聚吡咯凝胶作用下,电解反应转化率稍有所下降,但仍具有较高的反应选择性。因此,可以用廉价的碳棒来替代价格昂贵的金属电极。
[0066]
实施例20:
[0067]
在50ml无隔膜电解槽中加入0.069g p-meobzoh,0.0159g(5mmol/l)liclo4,0.03g过滤回收后的dbsa为掺杂剂的聚吡咯凝胶电解质,30ml溶剂,反应温度60℃,以pt作阳极,以pb作阴极,再加入6mm
×
10mm尺寸搅拌子,磁力搅拌设置为500rpm,启动直流电源控制电流为10ma,电解4h,得到目标产物对甲氧基苯甲醛。电解产物收率通过气相色谱gc进行分析,分析方法为面积归一法,产物收率如表4所示为。
[0068]
实施例21:
[0069]
反应步骤同实施例20,所不同的是电极材料分别以碳棒作为阴阳两极,进行上述恒电流电解实验,结果列于表4。
[0070]
表4回收的聚吡咯凝胶电解质作用下p-meobzoh的电化学转化性能
[0071][0072]
由上述实验结果可知,通过简单过滤回收的聚吡咯凝胶电解质仍具有良好的效果;表明聚吡咯凝胶电解质的回收利用是可行的。
[0073]
实施例22:间甲基苯甲醛(式(2-2))的制备:
[0074]
在50ml无隔膜电解槽中加入0.061g间甲基苯甲醇,0.0159g(5mmol/l)liclo4,0.03gdbsa掺杂的聚吡咯凝胶电解质,30ml溶剂(乙腈),反应温度60℃,以pt作阳极,以pb作阴极,再加入6mm
×
10mm尺寸搅拌子,磁力搅拌设置为500rpm,启动直流电源控制电流为
10ma,电解4h,得到目标产物间甲基苯甲醛。电解产物收率通过气相色谱gc进行分析,分析方法为面积归一法,产物收率98%。
[0075]
实施例23:对叔丁基苯甲醛(式(2-3))的制备:
[0076]
在50ml无隔膜电解槽中加入0.082g对叔丁基苯甲醇,0.0159g(5mmol/l)liclo4,0.03gdbsa掺杂的聚吡咯凝胶电解质,30ml溶剂(乙腈),反应温度60℃,以pt作阳极,以pb作阴极,再加入6mm
×
10mm尺寸搅拌子,磁力搅拌设置为500rpm,启动直流电源控制电流为10ma,电解4h,得到目标产物对叔丁基苯甲醛。电解产物收率通过气相色谱gc进行分析,分析方法为面积归一法,产物收率97%。
[0077]
实施例24:间氯苯甲醛(式(2-4))的制备:
[0078]
在50ml无隔膜电解槽中加入0.071g间氯苯甲醇,0.0159g(5mmol/l)liclo4,dbsa掺杂的聚吡咯凝胶电解质,30ml溶剂(乙腈),反应温度60℃,以pt作阳极,以pb作阴极,再加入6mm
×
10mm尺寸搅拌子,磁力搅拌设置为500rpm,启动直流电源控制电流为10ma,电解4h,得到目标产物间氯苯甲醛。电解产物收率通过气相色谱gc进行分析,分析方法为面积归一法,产物收率98%。
[0079]
实施例25:对三氟甲基苯甲醛(式(2-5))的制备:
[0080]
在50ml无隔膜电解槽中加入0.088g对三氟甲基苯甲醇,0.0159g(5mmol/l)liclo4,0.03g dbsa掺杂的聚吡咯凝胶电解质,30ml溶剂(乙腈),反应温度60℃,以pt作阳极,以pb作阴极,再加入6mm
×
10mm尺寸搅拌子,磁力搅拌设置为500rpm,启动直流电源控制电流为10ma,电解8h,得到目标产物对三氟甲基苯甲醛。电解产物收率通过气相色谱gc进行分析,分析方法为面积归一法,产物收率91%。
[0081]
本领域的技术人员容易理解,以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1