一种HPLC法分离测定曲安奈德和硝酸益康唑组合物中有关物质的方法与流程
本发明属于分析化学领域,具体涉及一种HPLC法分离测定曲安奈德和硝酸益康唑组合物中有关物质的方法。
背景技术:
曲安奈德为糖皮质激素,具有抗炎、止痒及抗过敏作用。硝酸益康唑为抗真菌药,对皮肤癣菌、霉菌和酵母菌(如念珠菌)等有抗菌活性,对某些革兰阳性菌也有效。曲安奈德和硝酸益康唑组合物临床可用于皮炎、湿疹、炎症性真菌性疾病等的治疗。
曲安奈德(Triamcinolone Acetonide)为中强效的氟化糖皮质激素,有甾体类化合物的母环,结构式如式Ⅰ所示。对酸、碱、温度和光照均较敏感,在特殊条件易降解,性质不稳定。
硝酸益康唑(Econazole Nitrate)为第一代咪唑类的硝酸盐,有咪唑环和醚类结构,结构式如式Ⅱ所示。其中咪唑环易被氧化,对氧较敏感,在特殊条件下也能降解。
曲安奈德和硝酸益康唑化学结构、理化性质差异大,在各种条件下的稳定性也不一致,两者制成复方制剂后,硝酸益康唑的酸性对曲安奈德的稳定性有 影响,样品存储中空气中的氧气对硝酸益康唑的稳定性有影响;再者,硝酸益康唑为弱碱性化合物,结构中共轭体系小,成盐后在紫外区的响应较弱,加上两个化合物紫外区吸收差异较大,根据《化学药物质量控制分析方法验证技术指导原则》项下要求,对杂质降解途径进行详细了解,为产品的存储、运输提供更好的、更合理的条件,研究制剂处方、工艺对两主药成分的相互影响,分别建立简单可行、灵敏度高、专属性好的有关物质质量控制方法势在必行,因此建立有关物质质量控制项,对产品有关物质进行控制,能提高产品的质量可控性和安全性。
各国药典均未收录有曲安奈德和硝酸益康唑单方制剂以及复方制剂中曲安奈德和硝酸益康唑及其有关物质的测定方法,Ch.P、EP、BP均只收录曲安奈德和硝酸益康唑原料药的有关物质测定方法。Ch.P2015年版中收载有曲安奈德益康唑乳膏,其项下无有关物质检测项,仅有含量的HPLC法。申请号为200510096394.6的发明专利中公开了曲安奈德益康唑凝胶及其制法和质量控制方法,该质量控制方法中的含量方法与中国药典2015年版曲安奈德益康唑乳膏的含量法一致,且也无有关物质检测项。均不能作为曲安奈德和硝酸益康唑组合物有关物质的控制,说明目前现有技术中公开的方法中无有效的曲安奈德和硝酸益康唑组合物有关物质的检测方法。
技术实现要素:
有鉴于此,本发明的目的是提供一种用于分离测定曲安奈德和硝酸益康唑组合物中有关物质的HPLC方法,该方法专属性强,灵敏度高,以保证曲安奈德和硝酸益康唑组合物的质量控制。
为实现上述目的,本发明的技术方案为:
一种HPLC法分离测定曲安奈德和硝酸益康唑组合物中有关物质的方法,采用反相液相色谱法;采用以十八烷基硅烷键合硅胶为填料的色谱柱,以磷酸氢二铵溶液和有机溶剂的混合液为流动相进行洗脱分离。
进一步,所述的方法,所述磷酸氢二铵溶液的浓度为0.005mol/L-0.02mol/L。
优选的,所述磷酸氢二铵溶液的浓度为0.01mol/L。
进一步,所述的方法,所述磷酸氢二铵溶液的pH值为5.5-6.5。优选的,所述磷酸氢二铵溶液的pH值为6.0。
进一步,所述的方法,所述磷酸氢二铵溶液选用磷酸、甲酸、乙酸、柠檬酸和三氟乙酸中的一种或多种进行pH调节。优选磷酸进行pH调节。
进一步,所述的方法,所述有机溶剂为乙腈和/或甲醇。
优选的,所述有机溶剂为乙腈。
进一步,所述的方法,所述十八烷基硅烷键合硅胶色谱柱填料的颗粒粒径为3-5μm。优选颗粒粒径为5μm。
进一步,所述的方法,选用色谱柱柱长为150-250mm。优选色谱柱柱长为150mm。
进一步,所述的方法,所述流动相的流速为0.5-1.5ml/min。
优选的,所述流动相的流速为1.0ml/min。
进一步,所述的方法,所述流动相采用线性梯度洗脱分离,线性梯度洗脱条件为:
0min:磷酸氢二铵溶液与有机溶剂的体积比为75-85:15-25;
50min:磷酸氢二铵溶液与有机溶剂的体积比为25-35:65-75;
60min:磷酸氢二铵溶液与有机溶剂的体积比为25-35:65-75;
62min:磷酸氢二铵溶液与有机溶剂的体积比为75-85:15-25;
65min:磷酸氢二铵溶液与有机溶剂的体积比为75-85:15-25。
优选的,所述流动相采用线性梯度洗脱分离,线性梯度洗脱条件为:
0min:磷酸氢二铵溶液与有机溶剂的体积比为80:20;
50min:磷酸氢二铵溶液与有机溶剂的体积比为30:70;
60min:磷酸氢二铵溶液与有机溶剂的体积比为30:70;
62min:磷酸氢二铵溶液与有机溶剂的体积比为80:20;
65min:磷酸氢二铵溶液与有机溶剂的体积比为80:20。
进一步,所述的方法,所述色谱柱柱温为25-45℃。优选的,色谱柱柱温为35℃。
进一步,所述的方法,洗脱分离后,再采用检测器进行测定,所述检测器选用紫外检测器、DAD检测器和VWD检测器中的任一种。
优选的,所述检测波长为225nm±5nm。
本发明还提供了一种HPLC法分离测定曲安奈德益康唑乳膏中曲安奈德和硝酸益康唑及有关物质的方法,包括如下进行的步骤:
取曲安奈德益康唑乳膏适量,加入乙腈,于60±5℃水浴中加热,振摇使溶解,然后置冰浴中放置30分钟以上,于每分钟4000转离心10分钟,取上清液为供试品溶液,取供试品溶液注入高效液相色谱仪,采用以十八烷基硅烷键合硅胶为填料的色谱柱,以磷酸氢二铵溶液和有机溶剂的混合液为流动相进行洗脱分离,并在分离后采用检测器对有关物质进行测定。
进一步,所述的一种HPLC法分离测定曲安奈德益康唑乳膏中曲安奈德和硝酸益康唑及有关物质的方法,所述的方法还包括以下步骤:
取曲安奈德、硝酸益康唑及有关物质对照品制备对照溶液,分别取对照溶液及供试品溶液进样,进行高效液相色谱分析,记录色谱图,按主成分自身对照法计算供试品中各已知杂质的含量,按主成分自身对照法计算供试品中其他单个杂质的含量,总杂质为各已知杂质与其他单个杂质之和。
如在本发明的一个具体实施例中公开了利用HPLC法分离测定曲安奈德益康唑乳膏中曲安奈德和硝酸益康唑及有关物质的方法,具体步骤如下:
色谱条件:色谱柱填料为十八烷基硅烷键合硅胶,柱长为150mm,检测器为DAD检测器,检测波长为225nm,以0.01mol/L磷酸氢二铵(用磷酸调解pH为6.0)和乙腈的混合液作为流动相进行梯度洗脱,洗脱比例为:0min:磷酸氢二铵溶液与有机溶剂的体积比为80:20;50min:磷酸氢二铵溶液与有机溶剂的体积比为30:70;60min:磷酸氢二铵溶液与有机溶剂的体积比为30:70;62min:磷酸氢二铵溶液与有机溶剂的体积比为80:20;65min:磷酸氢二铵溶 液与有机溶剂的体积比为80:20。流动相流速为1.0ml/min,柱温为35℃,进样体积为10μl。
(1)分别取羟苯甲酯和2,6-二叔丁基对甲基苯酚(BHT)各约10mg、曲安奈德20mg和硝酸益康唑50mg,置50ml量瓶中,加50%乙腈溶液超声使溶解,加入1ml用50%乙腈制成含杂质A、B、C各约0.2mg/ml的混合对照品溶液,并加30%乙腈稀释至刻度,摇匀,作为系统适用性试验溶液;
(2)取曲安奈德和硝酸益康唑组合物适量,用30%乙腈溶解,分别配制成每1ml含曲安奈德0.4mg和硝酸益康唑4mg的供试品溶液。
(3)精密量取供试品溶液0.5ml,置100ml量瓶中,用30%乙腈稀释至刻度,摇匀,作为对照溶液。
分别取系统适用性溶液、供试品溶液和对照溶液,注入液相色谱仪,按上述色谱条件进行测定,按带校正因子的主成分自身对照法计算供试品中各已知杂质的含量,按主成分自身对照法计算供试品中其他单个杂质的含量,总杂质为各已知杂质与其他单个杂质之和。
本发明的有益效果在于:
本发明提供的一种HPLC法分离测定曲安奈德和硝酸益康唑组合物中有关物质的方法,弥补了现有技术的空缺,用高效液相色谱法分离测定曲安奈德和硝酸益康唑组合物中曲安奈德和硝酸益康唑及有关物质,并能有效分离出辅料峰,使其不干扰有关物质的检测。该方法分离度好,专属性强,灵敏度高;且操作简单,具有简便、快速的优点,保证产品的安全有效,对于曲安奈德和硝酸益康唑组合物的质量控制有重要意义。
附图说明
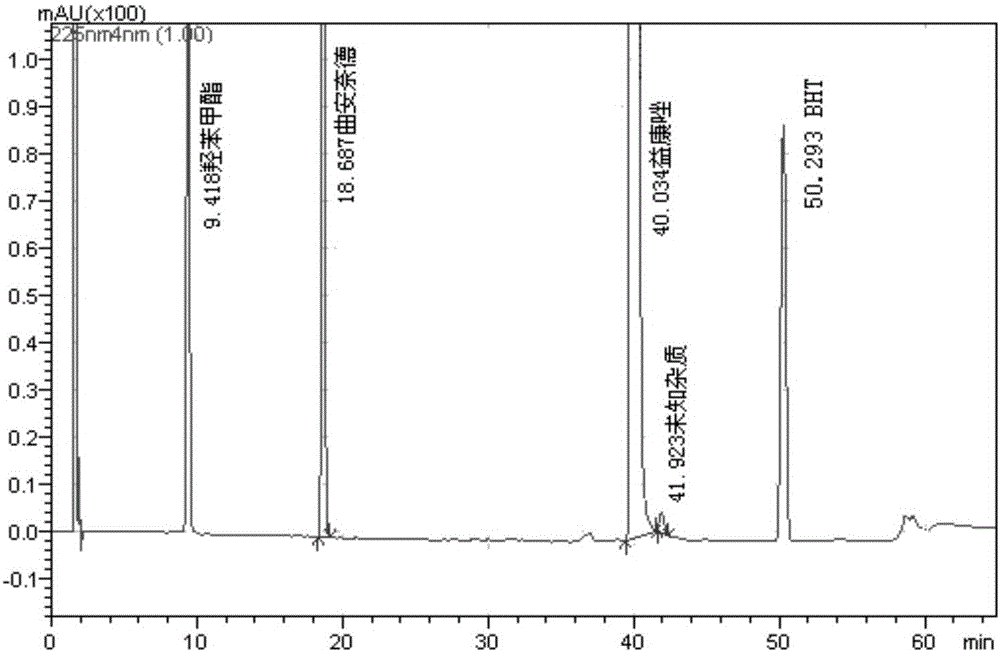
图1为实施例1中系统适用性溶液的HPLC图。
图2为实施例1中测定曲安奈德益康唑乳膏中的有关物质HPLC图。
图3为实施例1中测定空白辅料溶液的有关物质HPLC图。
图4为实施例1中测定溶剂的有关物质HPLC图。
具体实施方式
所举实施例是为了更好地对本发明的内容进行说明,但并不是本发明的内容仅限于所举实施例。所以熟悉本领域的技术人员根据上述发明内容对实施方案进行非本质的改进和调整,还包括各具体实施方式间的任意组合,仍属于本发明的保护范围。
实施例1曲安奈德益康唑乳膏中曲安奈德和硝酸益康唑及有关物质的测定
1、仪器与条件
仪器:高效液相色谱仪SHIMADZU 20A(SHIMADZU);
色谱工作站:LcSolution;
检测器:DAD;
色谱柱:十八烷基硅烷键合硅胶填充剂(SHIMADZU,VP-ODS,150mm×4.6mm,5μm);
流动相:流动相A:0.01mol/L磷酸氢二铵(用磷酸调节pH值为6.0);流动相B:乙腈;以流动相A、B进行线性梯度洗脱;
检测波长:225nm;
柱温:35℃;
流速:1.0ml/min;
进样体积:10μl。
流动相A、B线性梯度洗脱条件为:
2、实验步骤
(1)分别取羟苯甲酯和2,6-二叔丁基对甲基苯酚(BHT)各约10mg、曲安奈德20mg和硝酸益康唑50mg,置50ml量瓶中,加50%乙腈溶液超声使溶解,加入1ml用50%乙腈制成含杂质A、B、C各约0.2mg/ml的混合对照品溶液,并加溶剂(30%乙腈)稀释至刻度,摇匀,作为系统适用性溶液。
(2)取曲安奈德益康唑乳膏10g,置具塞锥形瓶中,加乙腈25ml,60℃水浴中加热,振摇使溶解,置冰浴中放置30分钟以上,于每分钟4000转离心10分钟,取上清液作为供试品溶液。
(3)另取本品曲安奈德益康唑乳膏的空白辅料10g,照供试品溶液相同的方法处理得空白辅料溶液。
(4)精密量取供试品溶液0.5ml,置100ml量瓶中,用溶剂(30%乙腈)稀释至刻度,摇匀,作为对照溶液。
(5)分别取系统适用性溶液、供试品溶液、空白辅料溶液和溶剂(30%乙腈),注入液相色谱仪,按上述色谱条件进行测定,记录色谱图,结果见图1-4。
3、检测结果
供试品溶液色谱图中如有杂质峰(保留时间为4-25min),杂质A、杂质B、杂质C的校正峰面积(峰面积分别乘以1.25、0.85、1.15)均不得大于对照溶液曲安奈德峰面积的2倍(1.0%),其他单个杂质峰面积不得大于对照溶液曲安奈德峰面积(0.5%),各杂质峰面积的和不得大于对照溶液曲安奈德峰面积的5倍(2.5%)。羟苯甲酯和峰面积小于对照溶液曲安奈德峰面积0.2倍的杂质峰均不积分(0.1%)。
上述供试品溶液色谱图中如有杂质峰(保留时间为25~50min),单个杂质的峰面积不得大于硝酸益康唑主峰面积(0.5%),各杂质峰面积的和不得大于硝酸益康唑主峰面积的2.0倍(1.0%)。BHT和峰面积小于对照溶液硝酸益康唑峰面积0.2倍的杂质峰均不积分(0.1%)。
按带校正因子的主成分自身对照法计算供试品中各已知杂质的含量,按主 成分自身对照法计算供试品中其他单个杂质的含量,总杂质为各已知杂质与其他单个杂质之和。
计算公式如下:
FAt/As
式中:At为供试品溶液中各杂质峰的峰面积;
As为对照溶液中曲安奈德或硝酸益康唑的主峰面积;
F为已知杂质的校正因子。
实施例2数批曲安奈德益康唑乳膏有关物质测定结果
按照实施例1的方法对我司数批曲安奈德益康唑乳膏样品进行有关物质测定。计算结果见表1。
表1数批样品检测结果
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。
- 还没有人留言评论。精彩留言会获得点赞!