冯了性风湿跌打药酒指纹图谱的建立方法及其指纹图谱与流程
本发明涉及冯了性风湿跌打药酒指纹图谱的建立方法,还涉及所述方法建立的冯了性风湿跌打药酒指纹图谱,属于冯了性风湿跌打药酒指纹图谱的建立领域。
背景技术:
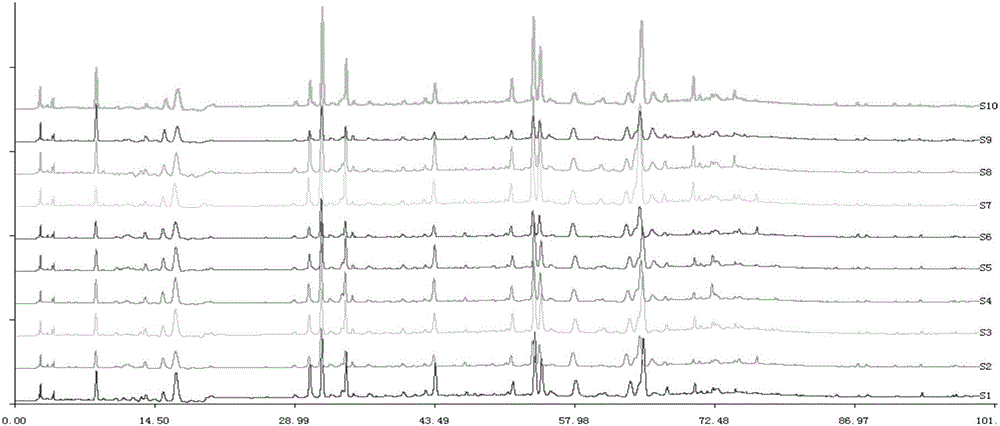
:冯了性风湿跌打药酒由丁公藤、麻黄、桂枝、枳壳、陈皮、黄精等27味中药材用白酒浸渍而成,可口服、可外用,具有祛风除湿,活血止痛的功效。用于风寒湿痹,手足麻木,腰腿酸痛;跌扑损伤,瘀滞肿痛等症。冯了性风湿跌打药酒的质量标准收载于自1990年版起的《中国药典》,《中国药典》2015版一部冯了性风湿跌打药酒项下的质量标准涵盖性状(东莨菪内酯、盐酸麻黄碱、厚朴酚、和厚朴酚、桂皮醛、去氢木香内酯、丹皮酚对照品及桂枝、木香对照药材)薄层鉴别,乙醇量、总固体、甲醇量的检查等检验指标,难以全面反映中药复方制剂的整体性和内在质量的全面控制。中药指纹图谱技术已成为国内外公认的鉴别中药品种和评价中药质量的最有效手段。指纹图谱是基于对中药物质群整体作用的认识,借助于波谱和色谱等技术获得中药化学成分的光谱或色谱图,是实现鉴别中药真实性、评价质量一致性和产品稳定性的可行模式。指纹图谱包括了对已知成分和未知成分的分析,反映的化学成分信息(具体表现为相对保留时间和相对峰面积)具有高度特异性和选择性,可较充分地反映出中药复杂混合体系中各种化学成分量分布的整体状况。色谱指纹图谱由于能够清晰地反映方剂中所含的主要成分情况,更具特征性和指纹性,因此各种色谱方法在方剂的指纹图谱质量控制研究中有着更普遍的应用。因此,建立一种冯了性风湿跌打药酒的指纹图谱,将为冯了性风湿跌打药酒的质量评价,为其生产和质量控制提供科学依据。技术实现要素:本发明所要解决的技术问题是提供一种冯了性风湿跌打药酒指纹图谱的建立方法,该方法简便易行、稳定可靠,所建立的冯了性风湿跌打药酒指纹图谱灵敏度高,稳定性好,能够用于冯了性风湿跌打药酒的质量控制。为解决上述技术问题,本发明所采取的技术方案是:本发明公开了一种冯了性风湿跌打药酒指纹图谱的建立方法,包括以下步骤:(1)将冯了性风湿跌打药酒稀释,过滤,收集滤液作为供试品溶液;(2)将供试品溶液进行高效液相色谱分析,得到HPLC指纹图谱;(3)根据HPLC指纹图谱中色谱峰的相对保留时间确定共有峰;(4)从共有峰中选择特征指纹峰,得到冯了性风湿跌打药酒指纹图谱。其中,步骤(2)所述高效液相色谱分析的条件包括:Agilent高效液相色谱仪;色谱柱为KromasilC18柱(规格为4.6×250mm,5μm)或EcosilC18柱(规格为4.6×250mm,5μm);流动相选自:按体积分数计,甲醇(A,即流动相A,下同)-0.1%醋酸:0.1%磷酸=1:1的水溶液(B,即流动相B,下同)、甲醇(A)-0.1%甲酸(B)、甲醇(A)-0.2%醋酸(B)、乙腈(A)-0.1%磷酸(B)或甲醇(A)-0.1%磷酸(B)中的任意一种;梯度洗脱;检测波长为210-360nm;流速1ml/min;柱温30℃;进样量5μl。作为本发明的优选技术方案,所述流动相为:按体积比计,甲醇(A)-0.1%醋酸:0.1%磷酸=1:1的水溶液(B)。所述梯度洗脱的步骤包括:按照体积分数计,0-20min,A为12%;20-30min,A由12%递升到25%;30-50min,A由25%递升到38%;50-60min,A为38%;60-75min,A由38%递升到60%;75-95min,A由60%递升到90%;90-100min,A为90%。本发明所述检测波长为210nm和300nm;优选的,所述检测波长为:0-10min为300nm,10-20min为210nm,20-100min为300nm。本发明以东莨菪苷、东莨菪内酯、绿原酸、盐酸麻黄碱、盐酸伪麻黄碱、桂皮醛、新橙皮苷、柚皮苷为参照物峰确定了冯了性风湿跌打药酒的HPLC指纹图谱。本发明方法通过比较10批供试品溶液的HPLC指纹图谱,根据色谱图中各色谱峰的相对保留时间,确定了15个共有峰为特征指纹峰;所述15个共有峰以绿原酸为参照的相对保留时间分别为:1号峰平均相对保留时间为0.265,RSD(相对标准偏差)<1%,优选的,RSD为0.37%,相对峰面积平均值为277.484;2号峰平均相对保留时间为0.494,RSD<1%,优选的,RSD为0.74%,相对峰面积平均值为203.291;3号峰平均相对保留时间为0.525,RSD<1%,优选的,RSD为0.61%,相对峰面积平均值为617.653;4号峰平均相对保留时间为0.960,RSD<1%,优选的,RSD为0.23%,相对峰面积平均值为288.293;5号峰平均相对保留时间为1.000,RSD<1%,优选的,RSD为0.2%,相对峰面积平均值为797.696;6号峰平均相对保留时间为1.078,RSD<1%,优选的,RSD为0.17%,相对峰面积平均值为276.629;7号峰平均相对保留时间为1.368,RSD<1%,优选的,RSD为0.15%,相对峰面积平均值为307.821;8号峰平均相对保留时间为1.621,RSD<1%,优选的,RSD为0.15%,相对峰面积平均值为212.11;9号峰平均相对保留时间为1.691,RSD<1%,优选的,RSD为0.16%,相对峰面积平均值为740.134;10号峰平均相对保留时间为1.713,RSD<1%,优选的,RSD为0.17%,相对峰面积平均值为496.374;11号峰平均相对保留时间为1.745,RSD<1%,优选的,RSD为0.18%,相对峰面积平均值为92.442;12号峰平均相对保留时间为1.824,RSD<1%,优选的,RSD为0.2%,相对峰面积平均值为370.97;13号峰平均相对保留时间为2.000,RSD<1%,优选的,RSD为0.28%,相对峰面积平均值为290.672;14号峰平均相对保留时间为2.047,RSD<1%,优选的,RSD为0.09%,相对峰面积平均值为1228.019;15号峰平均相对保留时间为2.081,RSD<1%,优选的,RSD为0.14%,相对峰面积平均值为194.893。本发明通过保留时间、紫外图谱比较以及对照品对照,从共有峰中指认出8个色谱峰,分别为盐酸麻黄碱(2,即共有峰编号,下同),盐酸伪麻黄碱(3),东莨菪苷(4),绿原酸(5),东莨菪内酯(7),柚皮苷(11),新橙皮苷(12)和桂皮醛(14)。本发明冯了性风湿跌打药酒指纹图谱的建立方法中,步骤(1)所述稀释为:用甲醇将冯了性风湿跌打药酒稀释1-3倍,优选为稀释1倍。步骤(1)所述过滤为过0.45μm微孔滤膜。本发明对冯了性风湿跌打药酒指纹图谱建立方法中的高效液相色谱分析的条件,包括检测波长、色谱柱和流动相进行了优化。检测波长的优化结果表明,用紫外DAD检测器对供试品溶液进行检测,在300nm条件下出峰最丰富,明显优于在246nm、274nm、340nm或360nm条件下出峰情况;但药酒中药材用量较大的麻黄的指标成分盐酸麻黄碱和盐酸伪麻黄碱在210nm处出峰。因此,本发明确定210和300nm作为检测波长。对于色谱柱的选择,本发明比较了3种不同品牌型号的色谱柱,从色谱峰峰形、分离度等方面比较,优选了EcosilC18柱(4.6×250mm,5μm)或KromasilC18柱(4.6×250mm,5μm)为试验色谱柱。流动相的优化结果表明,分别以甲醇-0.1%甲酸、甲醇-0.2%醋酸、乙腈-0.1%磷酸或甲醇-0.1%磷酸作为流动相,结果得出色谱峰均不理想;而选择甲醇-(0.1%醋酸:0.1%磷酸=1:1)作为流动相系统时,洗脱能力强,色谱图信息量大,色谱峰形好,分离效果好,基线平稳;在流动相中,磷酸增加紫外吸收值,有利于麻黄碱的检测;醋酸对其他峰有较好的分离效果。因此,本发明选择甲醇-(0.1%醋酸:0.1%磷酸=1:1)作为流动相系统。精密度试验表明,按照本发明指纹图谱的建立方法,取冯了性风湿跌打药酒平行进样6次并将结果导入“中药色谱指纹图谱相似度评价系统”进行色谱峰匹配,结果15个共有峰的相对保留时间RSD均<1%,相对峰面积RSD均<5%,表明精密度良好。稳定性试验表明,取冯了性风湿跌打药酒分别于0-24h进样,结果15个共有峰的相对保留时间RSD均<1%,相对峰面积RSD均<5%,表明本发明方法稳定性良好。重复性试验表明,将冯了性风湿跌打药酒供试品溶液6份分别进样,结果15个共有峰的相对保留时间RSD均<1%,相对峰面积RSD均<5%,表明本发明方法重复性良好。本发明进一步公开了所述方法建立的冯了性风湿跌打药酒指纹图谱,优选的,所述冯了性风湿跌打药酒指纹图谱如说明书附图图3所示。本发明所建立的冯了性风湿跌打药酒指纹图谱能够应用于冯了性风湿跌打药酒的质量控制或质量评价。指纹图谱的相似度评价结果表明,10批冯了性风湿跌打药酒指纹图谱的相似度基本在0.9以上,精密度良好。本发明技术方案与现有技术相比,具有以下有益效果:本发明采用HPLC-UV图谱分析建立了冯了性风湿跌打药酒的指纹图谱,该方法简便易行,精密度良好,重复性好,稳定可靠。本发明所建立的冯了性风湿跌打药酒指纹图谱灵敏度高、稳定性好,是一种有效的冯了性风湿跌打药酒质量评价方法,将为冯了性风湿跌打药酒的生产和质量控制提供科学依据。附图说明图1为冯了性风湿跌打药酒及对照品溶液的指纹图谱;其中,横坐标的单位为min;图2为冯了性风湿跌打药酒供试品的HPLC指纹图谱;其中,S1-S10为10批供试品;横坐标的单位为min;图3为冯了性风湿跌打药酒的指纹图谱;其中,1-15为特征指纹峰;横坐标的单位为min;图4为300nm作为检测波长的出峰情况;图5为246nm作为检测波长的出峰情况;图6为274nm作为检测波长的出峰情况;图7为340nm作为检测波长的出峰情况;图8为360nm作为检测波长的出峰情况;图9为210nm作为检测波长的出峰情况;图10为甲醇-0.1%甲酸为流动相的色谱峰;图11为甲醇-0.2%醋酸为流动相的色谱峰;图12为乙腈-0.1%磷酸为流动相的色谱峰;图13为甲醇-0.1%磷酸为流动相的色谱峰;图14为甲醇-(0.1%醋酸:0.1%磷酸=1:1)为流动相的色谱峰。具体实施方式下面结合具体实施例来进一步描述本发明,本发明的优点和特点将会随着描述而更为清楚。但是应理解所述实施例仅是范例性的,不对本发明的范围构成任何限制。本领域技术人员应该理解的是,在不偏离本发明的精神和范围下可以对本发明技术方案的细节和形式进行修改或替换,但这些修改或替换均落入本发明的保护范围。1、仪器与试药1.1仪器Agilent高效液相色谱仪(美国安捷伦公司,型号1260系列),电子天平(梅特勒-托利多,XS105DU)。1.2试药对照品东莨菪内酯(110768-200504)、桂皮醛(110710-201418)、绿原酸(110753-201314)、新橙皮苷(11857-201102)、盐酸麻黄碱(171241-201508)、盐酸伪麻黄碱(171237-201208)、柚皮苷(110722-201312)由中国药品生物制品检定所提供;东莨菪苷(纯度≥98%,批号Y24S6Y17687)由上海源叶生物科技有限公司提供。冯了性风湿跌打药酒(国药集团冯了性(佛山)药业有限公司生产,500mL/瓶)。甲醇为色谱纯,水为超纯水,其他试剂均为分析纯。实施例1冯了性风湿跌打药酒指纹图谱的建立1、实验方法1.1冯了性风湿跌打药酒指纹图谱的建立1.1.1色谱条件KromasilC18柱(4.6×250mm,5μm);流动相为(按体积比计)甲醇(A)-0.1%醋酸:0.1%磷酸=1:1的水溶液(B);梯度洗脱(按体积比计)(0~20min,A为12%;20~30min,A由12%递升到25%;30~50min,A由25%递升到38%;50~60min,A为38%;60~75min,A由38%递升到60%;75~95min,A由60%递升到90%;90~100min,A为90%);检测波长(0-10min为300nm,10-20min为210nm,20-100min为300nm);流速1ml/min;柱温30℃;进样量5μl。1.1.2供试品溶液的制备精密量取冯了性风湿跌打药酒10ml,置20ml量瓶中,加甲醇稀释至刻度,摇匀,过0.45μm微孔滤膜,取续滤液,即得。1.1.3对照品溶液的制备精密称取东莨菪内酯、桂皮醛、绿原酸、新橙皮苷、盐酸麻黄碱、盐酸伪麻黄碱、柚皮苷、东莨菪苷对照品适量,加80%甲醇水溶液溶解制成东莨菪内酯(40μg/ml)、桂皮醛(10μg/ml)、绿原酸(40μg/ml)、新橙皮苷(80μg/ml)、盐酸麻黄碱(40μg/ml)、盐酸伪麻黄碱(40μg/ml)、柚皮苷(80μg/ml)、东莨菪苷(212μg/ml)溶液,即得。1.2方法学考察1.2.1精密度试验取冯了性风湿跌打药酒,按照1.1.2的方法制备供试品溶液,按照1.1.1的色谱条件,平行进样6次,并将结果导入“中药色谱指纹图谱相似度评价系统”2004A版,进行色谱峰匹配。1.2.2稳定性试验取冯了性风湿跌打药酒,按照1.1.2的方法制备供试品溶液,按照1.1.1的色谱条件,分别于0,2,4,8,12,24h进样,考察本发明方法的稳定性。1.2.3重复性试验取冯了性风湿跌打药酒,按照1.1.2方法平行制备供试品溶液6份,按照1.1.1的色谱条件,分别进样,考察本发明方法的重复性。2、实验结果2.1指纹图谱的建立与分析2.1.1指纹图谱的建立本发明冯了性风湿跌打药酒供试品及对照品溶液的指纹图谱见图1。本发明按上述1.1.1的色谱条件进行测定,得到冯了性风湿跌打药酒S1-S10供试品的HPLC指纹图谱(图2),根据色谱图中各色谱峰的相对保留时间,确定共有峰,并选取15个共有峰为特征指纹峰(图3)。因图谱中绿原酸分离度好,出峰时间适中,峰面积较大且稳定,因此将其作为色谱参照峰S。所述15个共有峰以绿原酸为参照的相对保留时间见表1。表1S1-S10供试品HPLC指纹图谱15个共有峰的相对保留时间及相对峰面积本发明通过保留时间、紫外图谱比较以及对照品对照,从共有峰中指认出8个色谱峰,分别为盐酸麻黄碱(2,即共有峰编号,下同),盐酸伪麻黄碱(3),东莨菪苷(4),绿原酸(5),东莨菪内酯(7),柚皮苷(11),新橙皮苷(12)和桂皮醛(14)。2.1.2指纹图谱的相似度评价采用“中药色谱指纹图谱相似度评价软件”2004A版(国家药典委员会)对10批(S1-S10)冯了性风湿跌打药酒进行相似度评价,结果见表2。表210批冯了性风湿跌打药酒的相似度评价S1S2S3S4S5S6S7S8S9S10对照指纹图谱S11.0000.9300.9800.9400.9850.9290.9560.9740.9190.9550.988S20.9301.0000.9130.9280.9420.9550.8770.9120.9300.8970.945S30.9800.9131.0000.9050.9780.9050.9840.9710.9180.9720.988S40.9400.9280.9051.0000.9600.9690.8550.9190.9110.8930.944S50.9850.9420.9780.9601.0000.9520.9510.9710.9480.9600.992S60.9290.9550.9050.9690.9521.0000.8560.9010.9470.8810.941S70.9560.8770.9840.8550.9510.8561.0000.9690.8830.9810.973S80.9740.9120.9710.9190.9710.9010.9691.0000.9080.9860.987S90.9190.9300.9180.9110.9480.9470.8830.9081.0000.9100.945S100.9550.8970.9720.8930.9600.8810.9810.9860.9101.0000.983对照指纹图谱0.9880.9450.9880.9440.9920.9410.9730.9870.9450.9831.000结果表明,10批冯了性风湿跌打药酒指纹图谱的相似度基本在0.9以上,精密度良好。2.2方法学考察结果2.2.1精密度试验本发明取冯了性风湿跌打药酒平行进样6次,并将结果导入“中药色谱指纹图谱相似度评价系统"2004A版,进行色谱峰匹配,结果见表3。结果表明(表3,S1-S6为进样6次数据,为峰面积数据)15个共有峰(编号为1-15)的相对保留时间RSD均<1%,相对峰面积RSD均<5%,表明仪器稳定,精密度良好。表3精密度试验结果编号S1S2S3S4S5S6保留时间RSD(%)峰面积RSD(%)1192.374193.291193.069191.852193.184194.4330.330.46293.589100.384104.20999.920107.373104.5180.274.763139.954150.301145.430151.469137.297151.6200.774.254375.944382.082390.045418.230403.351419.6300.624.645107.001106.143115.885115.482111.398112.1040.273.696482.051484.085482.500485.005488.054488.1890.210.557143.608144.500144.181143.352146.476146.6420.140.998173.101173.284173.290173.633173.321180.8980.091.779124.141128.842129.468129.745131.670131.2880.042.0910354.968356.834356.662357.539358.435360.1010.030.4911288.593291.035291.139291.644293.448294.2520.030.6912351.148354.587352.411353.153356.274356.6650.060.6213238.089238.203240.456243.033245.486247.3320.081.5914669.048674.082669.737676.427680.011689.0670.071.1015180.151182.024182.786182.736183.487186.0280.071.052.2.2稳定性试验本发明取冯了性风湿跌打药酒,分别于0,2,4,8,12,24h进样(S1-S6),结果(表4,S1-S6为进样6次数据,为峰面积数据)15个共有峰(编号为1-15)的相对保留时间RSD均<1%,相对峰面积RSD均<5%,表明供试品溶液在24h内稳定,本发明方法的稳定性良好。表4稳定性试验结果编号S1S2S3S4S5S6保留时间RSD(%)峰面积RSD(%)1192.374193.291193.069191.852193.184194.4330.330.46293.589100.384104.20999.920107.373104.5180.274.763139.954150.301145.430151.469137.297151.6200.774.254375.944382.082390.045418.230403.351419.6300.624.645107.001106.143115.885115.482111.398112.1040.273.696482.051484.085482.500485.005488.054488.1890.210.557143.608144.500144.181143.352146.476146.6420.140.998173.101173.284173.290173.633173.321180.8980.091.779124.141128.842129.468129.745131.670131.2880.042.0910354.968356.834356.662357.539358.435360.1010.030.4911288.593291.035291.139291.644293.448294.2520.030.6912351.148354.587352.411353.153356.274356.6650.060.6213238.089238.203240.456243.033245.486247.3320.081.5914669.048674.082669.737676.427680.011689.0670.071.1015180.151182.024182.786182.736183.487186.0280.071.052.2.3重复性试验本发明取冯了性风湿跌打药酒,平行制备供试品溶液6份分别进样,结果(表5,S1-S6为进样6次的峰面积数据)15个共有峰(编号为1-15)的相对保留时间RSD均<1%,相对峰面积RSD均<5%,表明本发明方法重复性良好。表5重复性试验结果编号S1S2S3S4S5S6保留时间RSD(%)峰面积RSD(%)1195.046191.010191.779190.940192.281191.0120.220.822106.05697.71496.706104.481106.177106.6530.164.403184.365162.533176.416167.341165.031168.0820.314.814436.650448.090435.733442.124425.000389.6370.354.895154.575147.569149.428147.764148.062147.0760.131.886512.442484.259492.630484.762489.996485.3690.102.197149.017135.944138.233141.282143.137141.8380.073.178179.635172.963173.297172.152172.977172.0990.061.659136.853130.139133.249131.357132.167131.7930.051.7510374.508352.893360.368355.031358.385355.3020.052.1911305.819288.558294.755289.073293.395290.9160.052.1712370.906349.328355.070351.884355.312350.4020.072.2313256.257238.998242.693236.845242.585240.1120.112.8414712.222675.358683.473675.477676.044676.0270.132.1415182.448180.514182.165179.258181.691180.3540.080.68实验例1冯了性风湿跌打药酒指纹图谱建立方法的优化1、检测波长的选择本发明用紫外DAD检测器对供试品溶液(按照实施例1中1.1.2制备供试品溶液)进行检测,结果(图4)显示在300nm条件下出峰最丰富,明显优于在246nm、274nm、340nm、360nm条件下出峰情况(图5-图8);但药酒中药材用量较大的麻黄的指标成分盐酸麻黄碱和盐酸伪麻黄碱要在210nm处出峰(图9)。因此,本发明确定210和300nm作为检测波长。2、色谱柱的选择本发明比较了3根不同品牌型号的色谱柱,从色谱峰峰形、分离度等方面比较,优选了EcosilC18柱(4.6×250mm,5μm)、KromasilC18柱(4.6×250mm,5μm)为试验色谱柱。3、流动相的选择本发明在供试品溶液的制备及色谱条件同实施例1的条件下,分别选择了(体积百分比)甲醇-0.1%甲酸、甲醇-0.2%醋酸、乙腈-0.1%磷酸、甲醇-0.1%磷酸作为流动相进行试验,结果得出色谱峰均不理想(图10-图13);而选择甲醇-(0.1%醋酸:0.1%磷酸=1:1)作为流动相系统时,洗脱能力强,色谱图信息量大,色谱峰形好,分离效果好,基线平稳(图14、表6);流动相中,磷酸增加紫外吸收值,有利于麻黄碱的检测;醋酸对其他峰有较好的分离效果。因此,本发明选择甲醇-(0.1%醋酸:0.1%磷酸=1:1)作为流动相系统。表6甲醇-(0.1%醋酸:0.1%磷酸=1:1)作为流动相的色谱检测结果当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1