一种Pd2+检测试剂及其合成方法和应用与流程
本发明涉及Pd2+的检测技术,具体涉及(E)-4-(二苯基氨基)苯甲醛O-丙-2-炔-1-基肟化合物与甲烷(1:2)(TP1)及其合成方法,以及TP1作为试剂在检测Pd2+中的应用。
背景技术:
钯作为一种稀有的铂族元素,其在科学研究和工业生产中的重要意义,如牙科、燃料电池、不饱和烃加氢和珠宝首饰等,在科学研究和工业生产中也得到了广泛的关注,是航天、航空等高科技领域以及汽车制造业不可缺少的关键材料。然而,残留的钯往往会通过各种方式进入环境,包括土壤,植物和河流。在这种情况下,少量的钯可能通过皮肤吸收或食物和水的摄入而进入我们身体。它对人体健康有严重的危害,如导致过敏反应、严重的原发性皮肤病和眼部疾病等。主要原因是钯可以攻击细胞的生物大分子,如含有氨基酸、蛋白巯基、DNA等,这会干扰细胞的正常生理过程。
目前已报道的用于检测Pd2+的检测方法有原子吸收光谱法(AAS),高效液相色谱法与固相微萃取、电感耦合等离子体原子发射光谱(ICP/OES)和X射线荧光,通常需要昂贵的仪器和复杂的样品前处理,从而限制了它的应用。与它们相比,因为荧光标记法成本低廉,非破坏性和实时性的特点,已得到了广泛使用。基于此发展一种检测过程简单、灵敏、成本低、适合临床、科研使用的检测Pd2+的方法非常必要。
技术实现要素:
:
本发明的目的是提供一种Pd2+检测试剂及其合成方法,以及将该检测试剂用于定量检测Pd2+,而且检测时在水相中进行、选择性高、灵敏度高、速度快。
本发明提供的Pd2+检测试剂是(E)-4-(二苯基氨基)苯甲醛O-丙-2-炔-1-基肟化合物与甲烷(1:2)化合物(TP1),英文名:(E)-4-(diphenylamino)benzaldehyde O-prop-2-yn-1-yloxime compound with methane(1:2),结构式为:
(E)-4-(二苯基氨基)苯甲醛O-丙-2-炔-1-基肟化合物与甲烷(1:2)化合物的合成方法,步骤为:
(1)将三苯胺和氯化氧磷完全溶解于DMF中,搅拌;向混合物中倒入冰水,用氢氧化钠 中和,过滤;粗产物通过硅胶柱纯化,得到4-(N,N-二苯氨基)苯甲醛;
(2)向4-(N,N-二苯氨基)苯甲醛中加入盐酸羟胺和醋酸钠,搅拌,减压蒸馏,硅胶柱层析纯化,得到4-(N,N-二苯氨基)苯甲醛肟;
(3)将4-(N,N-二苯氨基)苯甲醛肟和氢化钠完全溶解于四氢呋喃中,搅拌,随后加入溴丙炔,搅拌,萃取,收集有机层,减压蒸馏和柱色谱分离得到淡黄色固体,即得到Pd2+检测试剂TP1。
本发明提供的一种快速检测Pd2+的方法,是基于TP1化合物,在pH为7.4的HEPES溶液中定量地检测Pd2+的含量;具体步骤为:
(1)、配制pH=7.4、浓度为10mM的HEPES缓冲溶液,配制2mM的TP1的乙醇溶液;
(2)、将2000μL pH 7.4的HEPES缓冲溶液和1μL TP1的乙醇溶液加到干净的荧光比色皿中,在荧光分光光度仪上检测,随着待测样的加入,423nm的荧光强度逐渐减弱;
(3)、将2000μL pH 7.4的HEPES缓冲溶液和1μL TP1的乙醇溶液加到另外七个荧光比色皿中,分别再加入体积为1.0、2.0、3.0、4.0、5.0、6.0、7.0μL的Pd2+溶液,在荧光分光光度仪上测定423nm对应的荧光强度F为7125、6429、5026、3833、2680、1835、1085、365,以Pd2+浓度为横坐标,以相对荧光强度F-F0为纵坐标绘制图,F0=7129,得到Pd2+浓度的工作曲线;线性回归方程为:F-F0=7129.5833-1014.9881c,c的单位为μM;
(4)、将2000μL pH 7.4的HEPES缓冲溶液和1μL TP1的乙醇溶液加到干净的荧光比色皿中,用微量进样器吸取VμL待测样品溶液,加入到此干净的荧光比色皿中,在荧光分光光度仪上检测,将测得的荧光强度代入步骤(3)的线性回归方程,得到浓度c,待测样品C待测样=2000μL×c×10-6/VμL,即可求得Pd2+的浓度。
该检测方法对Pd2+显示了高的灵敏性和选择性,检测过程在水相中进行,简便、快速,检测结果准确。
与现有技术相比,本发明具有如下优点和效果:1、检测体系成本低廉,合成简单;2、本发明的检测方法,在纯水相中进行,对Pd2+显示了高的灵敏性和选择性;3、检测手段简单,只需要借助荧光分光光度仪即可实现。
附图说明:
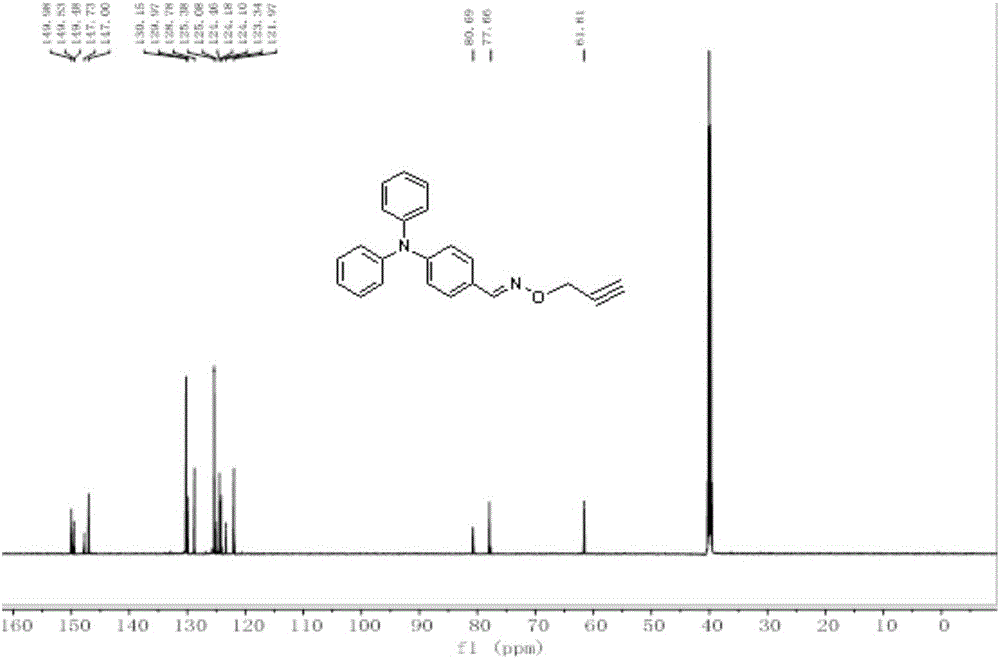
图1实施例1制备的TP1化合物的核磁氢谱图
图2实施例1制备的TP1化合物的核磁碳谱图
图3实施例1制备的TP1化合物的质谱图
图4实施例2TP1化合物与Pd2+作用的荧光发射图
图5实施例3TP1化合物与各种分析物的荧光柱状图
图6实施例4测定Pd2+的工作曲线
图7测定样品的荧光发射图
具体实施方式:
实施例1TP1化合物的制备及表征
将三苯胺(4.9g,0.005mol)和氯化氧磷(30g,0.2mol)完全溶解于50毫升DMF,1小时后,向混合物中倒入冰水,用20%的氢氧化钠中和后过滤;粗产物通过硅胶柱层析纯化(氯仿:石油醚=1:3作为洗脱剂),产率85%;
将4-(N,N-二苯氨基)苯甲醛(1.6克,6毫摩尔)溶解于二氯甲烷(25毫升)中,随后加入溶解于75ml乙醇中的盐酸羟胺(1.6克,24毫摩尔)和醋酸钠(2.4克,30毫摩尔),该混合物在室温下搅拌3小时,并减压蒸馏,粗产物经硅胶柱层析纯化(CH2Cl2作为洗脱剂)得到白色固体,产率85%;
将4-(N,N-二苯氨基)苯甲醛肟(46毫克,0.16毫摩尔)和氢化钠(8毫克,0.2毫摩尔)溶解于四氢呋喃(2毫升)并在0℃下搅拌30min;随后向混合物中加入1ml溶解于四氢呋喃的溴丙炔;搅拌1小时,在室温下,用水(10毫升)和乙酸乙酯(30毫升)萃取;收集有机层并用无水硫酸镁干燥,真空减压蒸馏和柱色谱层析纯化(正己烷/乙酸乙酯=2:1作为洗脱剂),得到淡黄色固体TP1化合物,产率:90%。
1H NMR(600MHz,25℃,CDCl3-d6):δ8.08(s,1H),7.47(d,2H,J=12,7.31(m,4H),7.25(m,6H),7.14(m,6H),4.77(d,2H),2.52(s,1H)(图1);13C NMR(150MHz,DMSO-d6):δ149.9,147.7,130.15,125.41,124.13,121.97,80.69,77.66,61.61(图2);calcd.for C22H18N2O[M+1]+:327.14,found 327.92(图3).
实施例2
配制pH=7.4、浓度为10mM的HEPES缓冲溶液,并用乙醇配制2mM的TP1化合物溶液;把2mL的HEPES(pH 7.4)溶液及1μL TP1化合物的乙醇溶液加到干净的荧光比色皿中,取Pd2+的溶液,逐渐用微量进样器加到此比色皿中,边加样边在荧光分光光度仪上检测,随着Pd2+的加入,423nm处荧光强度逐渐减弱,荧光发射图(见图4)。
实施例3
配制pH=7.4、浓度为10mM的HEPES缓冲溶液,并用乙醇配制2mM的TP1化合物溶液;在22个荧光比色皿中,各加入2mL的HEPES(pH 7.4)溶液和1μL的TP1化合物的乙醇溶液,再分别加入7摩尔当量Pd2+,以及10摩尔当量的各种分析物:Na+,Ni+,K+,Ag+,Au+,Zn2+,Ca2+,Mg2+,Cu2+,Pt2+,Mn2+,Fe3+,Cr3+and Al3+,在荧光分光光度仪上检测,绘制不同分析物对应的423nm相对荧光强度的柱状图,(见图5)。Pd2+使得TP1化合物的荧光淬灭,其 它的分析物没有引起TP1化合物荧光强度的变化。
经实验证明,其它分析物不干扰体系对Pd2+的测定。
实施例4
配制pH=7.4、浓度为10mM的HEPES缓冲溶液,并用乙醇配制2mM的TP1化合物溶液,用蒸馏水配制2mM的Pd2+溶液;把2mL的HEPES(pH 7.4)溶液和1μL的TP1化合物的乙醇溶液加到荧光比色皿中,分别再加入Pd2+溶液的体积为1.0、2.0、3.0、4.0、5.0、6.0、7.0时,在荧光分光光度仪上测定423nm对应的荧光强度F为7125、6429、5026、3833、2680、1835、1085、365,以Pd2+浓度为横坐标,以相对荧光强度F-F0为纵坐标绘制图,F0=7129,得到Pd2+浓度的工作曲线(见图6);线性回归方程为:F-F0=7129.5833-1014.9881c,c的单位为μM;
实施例5
配制pH=7.4的HEPES(10mM)缓冲溶液,配制2mM的Pd2+水溶液,并用乙醇配制2mM的TP1化合物溶液;把2mL的pH 7.4,HEPES溶液和1μL的TP1化合物的乙醇溶液加到干净的荧光比色皿中,取Pd2+的溶液6.5μL,用微量进样器加到此比色皿中,同时在荧光光谱仪上测定423nm的对应的荧光强度F为6784,通过实施例4的线性回归方程,求得c=7.37×10-6mol/L,偏差为0.87%。见图7。
- 还没有人留言评论。精彩留言会获得点赞!