七叶一枝花的鉴定方法与流程
本发明涉及中药材质量检测领域,具体涉及七叶一枝花的鉴定方法。
背景技术:
七叶一枝花为百合科植物p.polyphyllasmithvar.chinensis(franch.)hara.的干燥根茎,2015年版《中华人民共和国药典》规定以重楼皂苷为其质量评价标准,它与云南重楼均被药典规定为重楼的正规来源。重楼甾体皂苷是重楼的主要活性成分,具有抗肿瘤、消炎、止血、抗菌、镇痛、镇静、抗氧化、免疫调节、抑制血管生成、保护肝脏与肾脏作用。
云南白药集团、四川光大制药有限公司和通化万通药业股份有限公司等相关企业对重楼根茎资源需求量大,过度采挖野生资源,市场价格暴涨,目前重楼根茎野生资源日渐濒危,同时,人工种植目前尚无优质、稳定的种源供给市场,导致非药典重楼植物种的根茎充斥商品市场,已经成为制约重楼制药产业可持续发展的瓶颈,药材市场的重楼根茎很难采用常规的性状、显微、理化、薄层鉴别等方法进行准确的鉴别,因此,建立七叶一枝花的新鉴别方法显得特别必要。
技术实现要素:
本发明目的在于提供一种对七叶一枝花的单一和复合鉴定方法。
具体地,本发明提供了七叶一枝花的鉴定方法,它包括如下操作:
(1)取10批以上七叶一枝花,粉碎,压片后,采用红外光谱检测,测定区域4000-400cm-1,以各批样品检测数据中的各峰吸收强度平均值和各峰对应的波数为基础,得到红外光谱标准指纹图谱;
(2)取待检药材,采用步骤(1)相同的条件进行检测,获得待检药材指纹图谱;
(3)将待检药材指纹图谱与红外光谱标准指纹图谱进行相似度比较,鉴别待检药材的真伪;
其中,所述红外光谱标准指纹图谱中,至少在约3400cm-1、2940cm-1、1640cm-1、1411cm-1、1250cm-1、1200-950cm-1、1150cm-1处有吸收峰。
进一步地,所述红外光谱标准指纹图谱中,还在约1020cm-1、920cm-1、860cm-1、760cm-1、700cm-1处有吸收峰。
红外光谱图通常用波长(λ)或波数(σ)为横坐标,表示吸收峰的位置,用透光率(t%)或者吸光度(a)为纵坐标,表示吸收强度。
本发明一个具体实施方式中,所述红外光谱标准指纹图谱如图2所示。
其中,红外光谱检测前,选取七叶一枝花或待检药材与kbr的质量比为4:200进行压片。使用上述配比时,最强吸收峰的透光率在10以下,利于检测。
本发明实验表明,当相似度在0.90以上时,则可以判断待检药材为七叶一枝花。
另外,本发明还提供了七叶一枝花的另外一种鉴定方法,它至少包括采用红外光谱和x线粉末衍射两种鉴定方法,各方法鉴定次序不分先后,其中:红外光谱鉴定方法:(1)取10批以上七叶一枝花,粉碎,压片后,采用红外光谱检测,测定区域4000-400cm-1,以各批样品检测数据中的各峰吸收强度平均值和各峰对应的波数为基础,得到红外光谱标准指纹图谱;
(2)取待检药材,采用步骤(1)相同的条件进行检测,获得待检药材指纹图谱;
(3)将待检药材指纹图谱与红外光谱标准指纹图谱进行相似度比较,鉴别待检药材的真伪;
其中,所述红外光谱标准指纹图谱中,至少在约3400cm-1、2940cm-1、1640cm-1、1411cm-1、1250cm-1、1200-950cm-1、1150cm-1处有吸收峰;
x线粉末衍射鉴定方法:取10批以上七叶一枝花,采用x线粉末衍射进行检测,以各批样品检测数据中的各衍射峰峰强度平均值和各衍射峰对应的2θ值为基础,得到x线粉末衍射标准指纹图谱;取待检药材,采用前述相同的x线粉末衍射条件进行检测,获得待检药材指纹图谱;将待检药材指纹图谱与x线粉末衍射标准指纹图谱进行相似度比较;
当各鉴定方法中所得相似度均在0.90以上时,则判断待检药材为七叶一枝花。
本发明一个具体实施方式中,采用cukα辐射进行x线粉末衍射。
其中,扫描速度8°/min。
其中,狭缝宽度ds=ss=1°,rs=0.3mm。
其中,扫描范围2θ:5-65°。
其中,各样品进行检测前需要粉碎,并过100~200目筛。
本发明一个具体实施方式中,采用omnic软件处理红外光谱检测的指纹图谱;采用originpro软件处理x线粉末衍射检测的指纹图谱。另外,x线粉末衍射指纹图谱的数据处理,亦可采用专利201410105727.6中的相关方法。
本发明一个具体实施方式中,所述x线粉末衍射标准指纹图谱如图4所示。
进一步地,它还可以包括uplc鉴定方法:
取待检药材,采用甲醇或乙醇提取,提取物制备供试品溶液,经uplc检测;当色谱图中至少存在重楼皂苷vii和h相对应的色谱峰时,则判断待检药材为七叶一枝花;
其中,所述uplc色谱条件如下:
色谱柱:c18色谱柱,规格2.1mm×75mm,1.7μm
elsd条件:漂移管温度55℃,氮气压力40psi,增益500,冷却模式,柱温40℃,流速0.2ml·min-1
流动相为乙腈-水,梯度条件如下:
本发明研究过程中,采用3种不同的色谱柱behc18色谱柱(2.1mm×50mm,1.7μm)、hssc18色谱柱(2.1mm×75mm,1.7μm)、behc18色谱柱(2.1mm×100mm,1.7μm),试验结果表明:采用hssc18色谱柱(2.1mm×75mm,1.7μm)进行洗脱最佳,各色谱峰保留时间适中,峰形和分离均较好。
采用上述复合方法,可以更为精确地鉴定出七叶一枝花的真伪,为七叶一枝花的鉴定提供了新的方法。
本发明在实验中使用了相关系数法、向量夹角余弦法两种方法来计算相似度,两种方法均是相似度的常用方法,其计算公式如下:
本发明一个具体实施方式中,对于七叶一枝花而言,使用相关系数法进行相似度的判断,其判定结果可能优于向量夹角余弦法。
步骤(1)中药材批次的选取应保证在10批以上,但具体数值并不固定,只要满足统计学意义即可。另外,步骤(1)中所述“七叶一枝花或云南重楼”,是指经其他已知的成熟手段鉴定为七叶一枝花或云南重楼的正品。
步骤(2)中所述“待检药材”,可以是正品,也可以是伪品。本发明通过上述方法即可将正品与伪品鉴别区分。
指纹图谱是指某些复杂物质,比如中药,某种生物体或某种组织或细胞的dna,蛋白质经适当处理后,采用一定的分析手段,得到的能够标示其化学特征的色谱图或光谱图。“相似度”,已被国家药典委员会作为一个定性定量参数,确定为中药指纹图谱标准中的一项重要评价指标,目前被应用于色谱类指纹图谱的研究(国家药品监督管理局,2000)。
为了保证检测数据的准确性,可以按照常规方式对每个样品进行3次或以上的检测。
附图说明
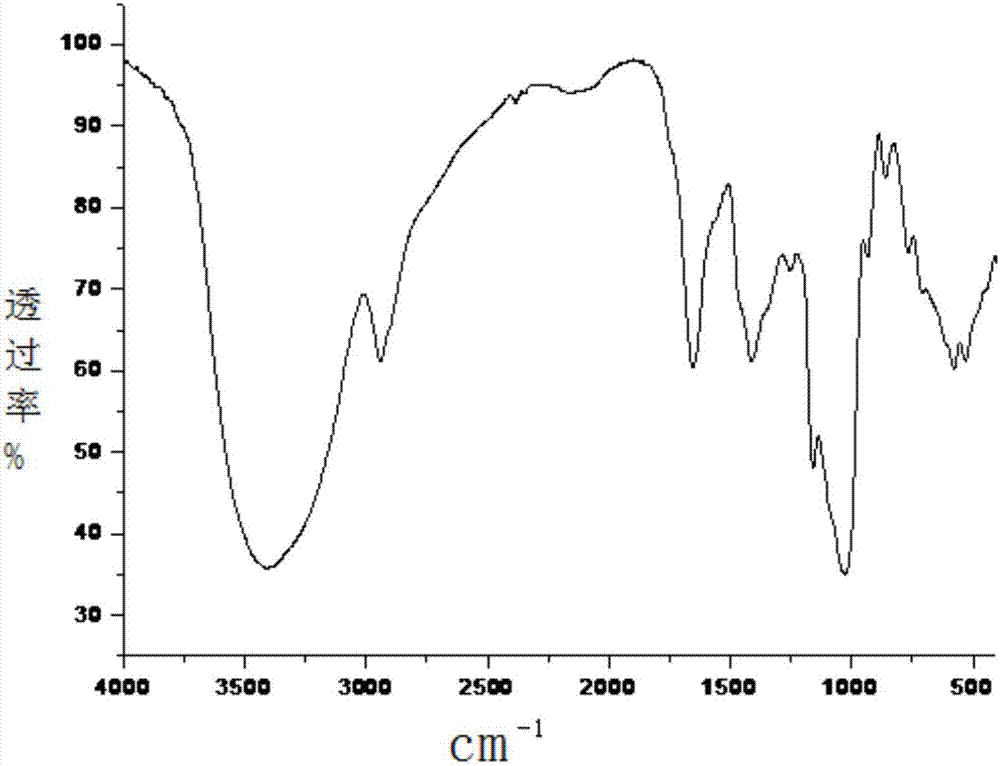
图1七叶一枝花根茎的红外图谱
图2七叶一枝花根茎的红外光谱标准指纹图谱
图3多批次七叶一枝花根茎的x射线衍射指纹图谱
图4七叶一枝花根茎的x射线衍射标准指纹图谱
图5混合对照品uplc色谱图,其中,1.重楼皂苷vii;3.重楼皂苷h;4.重楼皂苷vi;5.柴胡皂苷d;6.重楼皂苷ii;7.纤细薯蓣皂苷;8.重楼皂苷i
具体实施方式
仪器与材料
普析xd-6型x射线粉末衍射仪(北京普析通用仪器有限责任公司)、ir200傅立叶变换红外光谱仪(美国thermonecolet公司);hx-200型高速中药粉碎机(浙江永康溪岸五金药具厂);dhg-9246型电热恒温鼓风干燥箱(上海精宏实验设备有限公司)。玛瑙研钵。kbr粉末(光谱纯)(美国thermo公司)。
七叶一枝花及根茎由西南民族大学民族医药研究院刘圆教授、四川大学华西药学院张浩教授鉴定,原植物标本及药材样品保存在西南民族大学、四川大学华西药学院。样品信息见表1。
表1七叶一枝花样品基源及产地信息
实施例1红外指纹鉴定
检测方法:
样品处理:取原药材粉碎后,过5号筛备用。取各样品粉末60℃干燥6h。分别精取各样品4mg(使其最强吸收峰的透光率在10以下),加入kbr200mg,在红外灯下混合研磨均匀,放入模具内,压片,光谱分辨率4cm-1,测定区域4000-400cm-1,中速扫描,扫描次数16次,扫描时即时去除水分和co2的背景干扰为确保图谱的可比性。为了减少装样造成的误差,每个样品研磨均匀后平行扫描3次,即每次测定后将样品倒出重新装样压片,以保证光谱采集具有代表性,取3次测定得到的光谱平均值作为该样品的吸收光谱。
检测结果
进行光谱扫描,收集数据,并用红外光谱仪自带的ezomnic32应用软件来进行处理,扣除背景,并进行基线校正和自动平滑处理。得到各样品红外光图谱图(见图1)。
红外光谱标准指纹图谱的建立
在进行相似度分析之前,以10份七叶一枝花的根茎吸收强度的平均值为标准指纹图谱的吸收强度,最终得到的红外图谱如图2。
从图1、2可以观察到,其中3400cm-1附近强而宽的吸收峰,归属为多糖的羟基形成多种氢键o-h键、氨基n-h键的伸缩振动吸收;2940cm-1附近的吸收峰是多糖、脂类等亚甲基c-h反对称伸缩振动;1640cm-1附近的吸收峰主要为甾体苷、多糖类等物质中o-h弯曲振动和羰基c-o伸缩振动吸收峰;1411cm-1附近的吸收峰归属为亚甲基变角振动;1250cm-1附近吸收峰主要来自羧酸中c-oh的伸缩振动;在1200-950cm-1多糖吸收区显示了较强的吸收峰,主要为苷类、醇类或糖类物质c-o伸缩振动吸收峰;其中1150cm-1附近的吸收峰是多糖和苷类物质c-o键特征振动和甾体皂苷元中羟基o-h的弯曲振动。
另外,在1020cm-1附近吸收峰归属为直链c-c的伸缩振动;920、860、760、700cm-1附近的吸收峰为糖环中的c-c伸缩振动和甾体皂苷的特征吸收振动。
红外图谱相似度分析
以各样品的整个红外光谱图为分析对象,对样品的全谱进行相似度分析。把仪器测得数据点作为特征值,根据每个数据点位置所对应的吸收强度值的差异进行相似度分析。常用的相似度分析方法有“相关系数法”和“向量夹角余弦法”两种,计算公式如下:
计算各样品间相似度(相关系数和夹角余弦),计算结果见表1。
表2红外相似度计算结果
从表2中可知s8相对其他样品有一点差距,根据课题组采样时发现其栽培于土壤贫瘠、杂草丛生、日常管理疏忽的环境条件中;同时,前期试验得到的结果表明s8的皂苷总含量明显略低,这可能是引起差异变化的原因。用向量夹角余弦法计算相似度时,不同样品的计算结果极为接近,且与样品的质量状况没有明显的关系,不利于样品间的比较;而用相关系数法计算时,其结果则有较大的差异。经前期鉴定s8重楼皂苷含量远远低于药典规定0.6%,其相似度在0.92;其余红外图谱与对照图谱的相似度均在0.95以上。可见采用相关系数法对七叶一枝花的根茎红外指纹图谱进行全谱相似度分析时,其结果可以准确地反映出七叶一枝花根茎的实际质量状况,因此可用于七叶一枝花药材根茎的鉴别和质量评价。
实施例2七叶一枝花的复合鉴定方法
虽然目前实验中并没有发现与红外指纹图谱鉴定不符的案例,但是为了尽量避免药材鉴定中的疏漏,保证鉴定更高的准确性,本发明还使用了复合鉴定方法,即将红外、x线衍射等方法联合使用。
其中,红外鉴定方法如实施例1。
x线粉末衍射方法如下:
检测方法:
样品处理:取七叶一枝花根茎样品粉碎,60℃烘干6h,过100目筛备用。试验条件:cukα辐射;扫描方式:定性,步进扫描;电压/电流:36kv/20ma;扫描速度:8°/min;ds=ss=1°,rs=0.3mm;步宽:0.02°;扫描范围2θ:5-65°。
检测结果:
1、全部试验数据用originpro8.5处理(35点平滑),得到粉末衍射图谱,结果如图3。
2、x衍射标准指纹图谱的建立
在进行相似度分析之前,以10份七叶一枝花根茎峰强度的平均值为x衍射标准指纹图谱的峰强度,最终得到的x衍射标准指纹图谱如图4。
3、x衍射图谱相似度分析
常用的相似度分析方法有“相关系数法”和“向量夹角余弦法”两种,计算公式如下:
采用表格计算各样品间相似度(相关系数和夹角余弦)。
表3相似度计算结果
上述实施例根据峰的匹配结果采用平均值法计算获得x衍射标准指纹图谱的峰值;晶面间距d衍射相对强度i/i0表示衍射峰值;以角度2θ为横坐标,衍射峰值(d/i/i0)为纵坐标,形成了七叶一枝花根茎x衍射标准指纹图谱,并进行了相似度的比较。用向量夹角余弦法计算相似度时,s8与对照相似度较其他样品低,说明与样品的质量状况有一定关系,但是数据计算结果极其接近,差距不明显,不利于样品间的比较;而用相关系数法计算时,其结果则有较大的差异。经前期鉴定s8重楼皂苷含量远远低于药典规定0.6%,其相似度在0.9987,这一分析结果与红外光谱鉴别结果一致;其余x衍射图谱与标准指纹图谱的相似度均在0.9992以上。根据比较的结果可见相似度均大于0.9900,体现了彼此之间非常高的相似性,进一步说明了四川产区的不同七叶一支花根茎质量的相对稳定性,同时也说明了建立的x衍射标准指纹图谱可靠。
实施例3七叶一枝花的复合鉴定方法
除了实施例2中将红外与x线粉末衍射结合鉴定的方法外,本发明中还可以加上uplc。
红外和x线粉末衍射鉴定方法如实施例2。
uplc鉴定方法如下:
色谱条件
acquity
表4梯度洗脱程序
供试品溶液的制备
将干燥的七叶一枝花根茎粉碎,过40目筛,精密称取重楼根茎粉末0.500g,置于100ml锥形瓶中,按料液比1:100(g:ml)加入50ml甲醇,摇匀,静置4h后,超声30min,过滤,取滤液置于烧瓶,重复上述操作1次。合并滤液,45℃减压旋蒸至干,残渣用色谱甲醇溶解后,定容于10ml容量瓶,经0.22μm微孔滤膜过滤,备用。
对照品溶液的制备
外标法对照品溶液的制备
分别精密称取各重楼皂苷对照品,用色谱甲醇溶解,各对照品溶液中对照品浓度分别为0.206(vii)、0.210(h)、0.206(vi)、0.208(ii)、0.204(纤细薯蓣皂苷)、0.202(i)mg·ml-1,即得各重楼对照品溶液,经0.22μm微孔滤膜过滤,备用。
内标法对照品溶液的制备
分别精密称取各重楼皂苷对照品,用色谱甲醇溶解,各对照品溶液中对照品浓度分别为0.136(vii)、0.113(h)、0.104(vi)、0.121(ii)、0.106(纤细薯蓣皂苷)、0.103(i)mg·ml-1,即得各重楼对照品溶液,经0.22μm微孔滤膜过滤,备用。精密称取柴胡皂苷d,用色谱甲醇溶解,对照品浓度为0.250(柴胡皂苷d)mg·ml-1,即得柴胡皂苷d对照品溶液。
方法学考察
线性关系考察
外标法:精密吸取对照品溶液适量均稀释到2ml,配制成不同稀释倍数的混合对照品溶液,进样2μl,按色谱条件测定。以峰面积为纵坐标(y),进样量(μg)为横坐标(x),绘制外标法标准曲线,结果见表5。
表5外标法对照品的线性回归方程
内标法:精密吸取各重楼皂苷对照品溶液0.5,1,1.5,2.5,3ml,分别精密加入内标物(柴胡皂苷d)溶液1ml,定容至4ml,即得系列测定用对照品溶液,进样2μl,按前述色谱条件测定。以内标物(柴胡皂苷d)与对照品重楼皂苷的峰面积比值为纵坐标(y),它们的进样量比值为横坐标(x),绘制内标法标准曲线,结果见表6。
表6内标法对照品的线性回归方程
精密度试验
取对照品溶液,精密吸取,连续进样5次,测定记录,得到重楼皂苷vii、h、vi、ii、纤细薯蓣皂苷、i峰面积的rsd分别为1.14%、1.36%、1.28%、1.77%、1.81%、1.55%,结果<3%,表明仪器精密度好。
重复性试验
取样品s3,平行制备5份根茎供试品溶液及内标法系列供试品溶液,按前述色谱条件测定,记录各重楼皂苷峰面积,计算rsd,结果均<3%,表明方法重复性好。
稳定性试验
取同一供试品溶液(s3),按前述色谱条件,分别于0、2、4、8、12、24h进样测定,记录各重楼皂苷峰面积,计算rsd,结果均<3%,表明在24h内重楼供试品溶液稳定性好。
加样回收试验
取同一批已知含量的重楼药材样品(s3)5份,分别加精密称量加入重楼皂苷vii、h、vi、ii、纤细薯蓣皂苷、i对照品,制样,定容,测定,计算回收率。按外标法计算重楼皂苷vii、h、vi、ii、纤细薯蓣皂苷、i平均加样回收率分别为:99.91%、99.27%、98.44%、101.33%、98.78%、99.01%,rsd为1.76%、1.65%、1.44%、1.98%、1.52%、1.73%;按内标法计算,重楼皂苷vii、h、vi、ii、纤细薯蓣皂苷、i平均加样回收率分别为:97.21%、98.56%、100.12%、100.15%、99.34%、98.78%,rsd为0.92%、1.77%、1.81%、1.65%、1.34%、1.80%。
检测结果
表710份样品根茎中6个甾体皂苷含量(%)
检测的10批样品中,药典检测指标之一的重楼皂苷vii在所有样品中均检测到,而重楼皂苷h在所有样品中也都检测到。其余皂苷成分并非全都存在于各样品中,因此,本发明认为,可以同时以重楼皂苷vii和h作为鉴定七叶一枝花真伪的对照成分,若以此uplc方法结合红外和x线粉末衍射,将会更有利于提高七叶一枝花真伪鉴定的准确性。
- 还没有人留言评论。精彩留言会获得点赞!