一种奥贝胆酸有关物质的检测方法与流程
本发明涉及一种奥贝胆酸有关物质的检测方法。
背景技术:
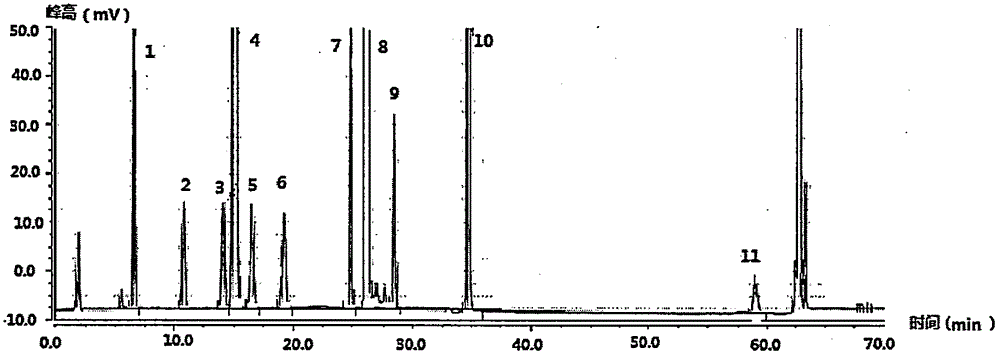
:药品是一类特殊商品,是用来诊断、预防及治疗疾病的一类特殊物质,与人们的健康和生命安危有极其密切的关系,其质量控制极为重要。药物安全性评价的一个重要内容是药物中有关物质的质量分数控制。在药物分析中所称的“有关物质”是指药物中的有机杂质,主要来源于药物活性成分的制备过程,如起始原料、试剂、中间体、副产物和异构体等物质,也可能是制剂在生产、贮藏和运输过程中产生的降解产物、聚合物或晶型转变过程等特征杂质。有关物质的种类与药物的合成路线和制剂工艺密切相关,通过对这些有关物质的测定,可以设法改变其合成路线、实验条件等因素,避免产生有关物质或使其降到最低限度。奥贝胆酸(obeticholicacid),又名int-747,由美国intercept制药公司研制开发,适应症为原发性胆汁性肝硬化(pbc)和非酒精性脂肪性肝病(nash),该产品已在美国上市。奥贝胆酸是一种半合成的鹅去氧胆酸,也是法尼酯衍生物x受体的特异性激动剂,是二十年来首个研发用于治疗胆汁淤积性肝病的药物,市场潜力巨大。奥贝胆酸,化学名为6α-乙基-3α,7α-二羟基-5β-胆烷酸,其结构式如下:奥贝胆酸的有关物质根据合成方法的不同而有差异,目前合成奥贝胆酸的方法有以下几种:专利wo02072598及文献journalofmedicinalchemistry,2002,45(17):3569-3572于2002年首次公开报道了包含奥贝胆酸在内的鹅去氧胆酸衍生物的制备方法,由于反应条件苛刻,试剂的毒性较大,在生产中较少采用;原研公司的专利wo2006122977(中国同族专利cn101203526)公开的方法是以7-klca为起始原料,分别经过c-24羧酸酯化、3-羟基硅醚保护、7-羰基形成硅烯醇醚、与乙醛羟醛缩合、c-24酯水解、6-亚乙基氢化、6-乙基构型转化和7-羰基的选择性还原八步反应,合成路线如下所示:原研公司的专利cn104781272a提及的检测中间体杂质的hplc方法对上述路线中的有关物质进行分析,发现图谱部分杂质特征峰无法分离,甚至重合。专利申请cn106749466a公开了的合成工艺如下:合成路线3中的奥贝胆酸的有关物质如下:ob-6、ob-a、ob-b、ob-c、ob-d、ob-e、ob-f、ob-g、ob-h、ob-i,其中ob-f为鹅去氧胆酸,为奥贝胆酸合成工艺的起始物料;ob-a、ob-b、ob-c是奥贝胆酸立体异构体;ob-6是合成工艺中间体杂质,为工艺杂质,ob-d、ob-e、ob-h、ob-i也为工艺合成过程中产生的工艺杂质,ob-g为降解杂质,上述杂质结构见表0:表0奥贝胆酸各杂质名称及结构目前,现有的奥贝胆酸有关物质的检测方法的分离效果不佳。因此,本领域亟需一种分析效果好的奥贝胆酸有关物质的检测方法。技术实现要素:本发明所要解决的技术问题是现有的奥贝胆酸有关物质的检测方法的分离效果不佳等缺陷,故而,本发明提供了一种奥贝胆酸有关物质的检测方法,该方法快速、有效、分离度好、操作简单、成本低,解决了奥贝胆酸特征峰与其异构体、起始物料、关键中间体、工艺杂质及降解杂质特征峰分离和测定的问题。本发明提供了一种奥贝胆酸有关物质的检测方法,其包括下述步骤:采用高效液相色谱法,将待测物在c18柱中进行梯度洗脱,即可;所述的待测物含有奥贝胆酸、奥贝胆酸盐和奥贝胆酸有关物质中的一种或多种;所述的梯度洗脱所使用的流动相为流动相a、流动相b和流动相c中的一种或多种;所述的流动相a为酸的水溶液;所述的流动相b为乙腈;所述的流动相c为甲醇;所述的梯度洗脱为三段式梯度洗脱或四段式梯度洗脱;当所述的梯度洗脱为三段式梯度洗脱时,其第一梯度中,流动相a的体积比例(其是指流动相a的体积占流动相总体积的比例,其余的体积比例均同此)为25%~40%(例如32%)、流动相b的体积比例为50%、流动相c的体积比例为10%~25%(例如18%);其第二梯度中,流动相a的体积比例为0%~5%、流动相b的体积比例为95%~100%、流动相c的体积比例为0%;其第三梯度中,流动相a的体积比例为25%~40%(例如32%)、流动相b的体积比例为45%~50%、流动相c的体积比例为15%~25%(例如18%);当所述的梯度洗脱为四段式梯度洗脱时,其第一梯度中,流动相a的体积比例为25%~40%(例如34%)、流动相b的体积比例为50%、流动相c的体积比例为10%~25%(例如16%);其第二梯度中,流动相a的体积比例为10%~30%(例如20%)、流动相b的体积比例为60%~70%、流动相c的体积比例为10%~20%;其第三梯度中,流动相a的体积比例为0%~5%、流动相b的体积比例为95%~100%、流动相c的体积比例为0%;其第四梯度中,流动相a的体积比例为25%~40%(例如34%)、流动相b的体积比例为46%~50%、流动相c的体积比例为14%~25%(例如16%)。在所述的检测方法中,所述的待测物可含有奥贝胆酸有关物质、以及、奥贝胆酸和/或奥贝胆酸盐。在所述的检测方法中,所述的奥贝胆酸盐中的“盐”为药学上可接受的有机或无机盐,例如钙盐、钠盐、钾盐、镁盐、盐酸盐、磷酸盐、硝酸盐、甲酸盐、醋酸盐、乙二酸盐、丙酸盐、酒石酸盐、硫酸盐、硫酸氢盐、碳酸盐、碳酸氢盐、苯甲酸盐、枸橼酸盐和甲磺酸盐中的一种或多种。在所述的检测方法中,所述的奥贝胆酸有关物质为本领域奥贝胆酸的有关物质,例如下述结构中的一种或多种:在所述的检测方法中,所述的待测物还可以含有无机杂质。在所述的检测方法中,所述的待测物还可以含有残留溶剂。在所述的检测方法中,所述的待测物还可以含有药用辅料。在所述的检测方法中,所述的待测物可以包括所述的奥贝胆酸、所述的奥贝胆酸有关物质、所述的无机杂质和所述的残留溶剂,例如由它们组成(又例如奥贝胆酸原料药)。所述的奥贝胆酸原料药中,所述的奥贝胆酸的质量百分比可为97~98%,例如97.8%。所述的奥贝胆酸原料药可为本领域常规的奥贝胆酸原料药,例如按照cn106749466a记载的方法制备的奥贝胆酸原料药。所述的cn106749466a记载的方法可包括下述步骤:(1)催化加氢反应,化合物vi在催化剂作用下催化氢化反应得到化合物vii;(2)构型转化,化合物vii在非亲核性强碱或酸的作用下转化构型得到化合物vii’;(3)还原反应,化合物vii’在还原剂作用下,发生羰基还原反应得到化合物viii;(4)水解反应,在碱性条件下,化合物viii经过酯水解得到化合物i;在所述的步骤(1)中,所述的催化剂可选自pd/c、pto2、pt/c、ru/c、rh/c和兰尼镍中的一种或多种。在所述的步骤(1)中,所述的催化加氢反应的溶剂可选自乙酸、甲醇、乙醇、四氢呋喃和乙酸乙酯中的一种或多种。在所述的步骤(1)中,所述的催化加氢反应可加入盐酸或醋酸钠调节溶剂的ph。在所述的步骤(2)中,所述的非亲核性强碱可选自三乙胺、甲醇钠、乙醇钠和dbu中的一种或多种。在所述的步骤(2)中,所述的酸可选自盐酸、三甲基氯硅烷、甲磺酸和对甲苯磺酸中的一种或多种。在所述的步骤(2)中,所述的构型转化反应的溶剂可为无水甲醇、无水乙醇和四氢呋喃中的一种或多种。在所述的步骤(2)中,所述的构型转化反应的温度可在室温与溶剂回流温度之间。在所述的步骤(3)中,所述的还原剂可为硼氢化钾或硼氢化钠,相应的还原反应的溶剂可为甲醇、乙醇、四氢呋喃或者甲醇/水、乙醇/水、或、四氢呋喃/水的混合溶液。在所述的步骤(3)中,所述的还原剂可为乙硼烷,相应的还原反应的溶剂可为无水四氢呋喃或者无水乙醚。在所述的步骤(4)中,所述的碱可为氢氧化钾、氢氧化钠或者氢氧化锂。在所述的步骤(4)中,所述的酯水解反应的溶剂可为甲醇、乙醇、四氢呋喃或者甲醇/水、乙醇/水、四氢呋喃/水的混合溶液。在所述的检测方法中,所述的待测物可以包括所述的奥贝胆酸盐、所述的奥贝胆酸有关物质、所述的无机杂质和所述的残留溶剂,例如由它们组成(又例如奥贝胆酸盐原料药)。在所述的检测方法中,所述的待测物可以包括所述的奥贝胆酸、所述的奥贝胆酸盐、所述的奥贝胆酸有关物质、所述的无机杂质和所述的残留溶剂,例如由它们组成。在所述的检测方法中,所述的待测物可以包括所述的奥贝胆酸、所述的奥贝胆酸有关物质、所述的无机杂质、所述的残留溶剂和所述的药用辅料,例如由它们组成(又例如奥贝胆酸制剂)。所述的奥贝胆酸制剂的剂型可为本领域常规的剂型,例如片剂、胶囊剂、混悬液、膜剂、颗粒剂、口服液或注射剂。在所述的检测方法中,所述的待测物可以包括所述的奥贝胆酸盐、所述的奥贝胆酸有关物质、所述的无机杂质、所述的残留溶剂和所述的药用辅料,例如由它们组成(又例如奥贝胆酸盐制剂)。在所述的检测方法中,所述的待测物可以包括所述的奥贝胆酸、所述的奥贝胆酸盐、所述的奥贝胆酸有关物质、所述的无机杂质、所述的残留溶剂和所述的药用辅料,例如由它们组成。在所述的检测方法中,所述的待测物在梯度洗脱前可进行预处理,以符合进样标准。所述的预处理可为本领域常规的预处理,例如,当所述的待测物为所述的奥贝胆酸原料药时,所述的预处理包括下述步骤:将所述的待测物溶解于溶剂中,所述的溶剂可为甲醇、乙腈、甲醇和水的混合溶剂、乙腈和水的混合溶剂、或者、“水-甲醇-乙腈”混合溶剂(例如体积比为36∶14∶50或32∶18∶50的“水-甲醇-乙腈”混合溶剂)。在所述的检测方法中,所述的c18柱的填料可为十八烷基硅烷键合硅胶。在所述的检测方法中,所述的c18柱的柱长可为本领域常规的柱长,例如100mm~300mm,又例如250mm。在所述的检测方法中,所述的c18柱的内径可为本领域常规的内径,例如1mm~10mm,又例如4.6mm。在所述的检测方法中,所述的c18柱的粒径可为本领域常规的粒径,例如1μm~10μm,又例如5μm。在所述的检测方法中,所述的c18柱可为c18(4.6mm×250mm,5μm)的十八烷基硅烷键合硅胶色谱柱。在所述的检测方法中,所述的c18柱的柱温可为本领域常规的柱温,例如25~40℃,又例如30℃。在所述的检测方法中,所述的流动相a中的酸可为挥发性酸。所述的挥发性酸可为本领域常规的挥发性酸,例如三氟乙酸、甲酸、乙酸和碳酸中的一种或多种,又例如三氟乙酸。在所述的检测方法中,所述的流动相a的ph可为本领域常规的ph,例如2.0~4.0,又例如2.2。在所述的检测方法中,所述的流动相a可为ph为2.2的三氟乙酸水溶液。在所述的检测方法中,当所述的梯度洗脱为三段式梯度洗脱时,各梯度的洗脱时间可有本领域技术人员视具体情形调整(不会付出创造性劳动),例如:其第一梯度的时间段可为0min~37min;其第二梯度的时间段可为38min~64min;其第三梯度的时间段可为65min~70min。在所述的检测方法中,当所述的梯度洗脱为四段式梯度洗脱时,各梯度的洗脱时间可有本领域技术人员视具体情形调整(不会付出创造性劳动),例如:其第一梯度的时间段可为0min~20min;其第二梯度的时间段可为21min~30min;其第三梯度的时间段可为31min~60min;其第四梯度的时间段可为61min~70min。在所述的检测方法中,所述的梯度洗脱的流速可为本领域常规的流速,例如0.8-1.3ml/min,又例如1.2ml/min。在所述的检测方法中,所述的梯度洗脱的进样量可为本领域常规的进样量,例如10~100μl。在所述的检测方法中,所述的梯度洗脱的运行时间可为本领域常规的运行时间,例如40-80min,又例如70min。在所述的检测方法中,所述的检测方法还可包括下述步骤:在梯度洗脱后,采用蒸发光散射检测器检测洗脱液。在所述的检测方法中,所述的蒸发光散射检测器可为本领域常规的蒸发光散射检测器,例如alltech-6000。在所述的检测方法中,所述的蒸发光散射检测器的漂移管的温度可为本领域常规的温度,例如90~100℃,又例如95℃。在所述的检测方法中,所述的蒸发光散射检测器的气体流速可为本领域常规的气体流速,例如2.0-2.8l/min,又例如2.5l/min。如无特别说明,本发明中所用的术语具有以下含义:“有关物质”包括工艺中引入的杂质和降解产物等,可能是已知的或未知的、挥发性的或不挥发性的。其化学结构一般与活性成分类似或具渊源关系。“无机杂质”是指在原料药及制剂生产或传递过程中产生的杂质,这些杂质通常是已知的,主要包括:反应试剂、配位体、催化剂、重金属、其它残留的金属、无机盐、助滤剂、活性炭等。“残留溶剂”是指在原料药及制剂生产过程中使用的有机溶剂,其研究可参考有机溶剂残留量研究的技术指导原则。“药用辅料”可为药物生产领域中广泛采用的那些辅料。辅料主要用于提供一个安全、稳定和功能性的药物组合物,还可以提供方法,使受试者接受给药后活性成分以所期望速率溶出,或促进受试者接受组合物给药后活性成分得到有效吸收。所述的药用辅料可以是惰性填充剂,或者提供某种功能,例如稳定该组合物的整体ph值或防止组合物活性成分的降解。所述的药用辅料可以包括下列辅料中的一种或多种:粘合剂、助悬剂、乳化剂、稀释剂、填充剂、成粒剂、胶粘剂、崩解剂、润滑剂、抗粘着剂、助流剂、润湿剂、胶凝剂、吸收延迟剂、溶解抑制剂、增强剂、吸附剂、缓冲剂、螯合剂、防腐剂、着色剂、矫味剂和甜味剂。在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。本发明所用试剂和原料均市售可得。本发明的积极进步效果在于:本发明的检测方法能够有效的测定奥贝胆酸溶液中各种相关杂质的存在,杂质分离度均大于1.5,不对称度均在0.88~1.2之间,理论塔板数均在10000以上。附图说明图1为按照实施例1的条件分析系统适用性溶液得到的hplc图。图2为按照实施例2的条件分析系统适用性溶液得到的hplc图。图3为按照实施例3的条件分析系统适用性溶液得到的hplc图。图4为按照实施例4的条件分析系统适用性溶液得到的hplc图。图5为按照实施例5的条件分析系统适用性溶液得到的hplc图。图6为按照实施例6的条件分析系统适用性溶液得到的hplc图。图7为按照对比例1的条件分析系统适用性溶液得到的hplc图。图8为按照对比例2的条件分析系统适用性溶液得到的hplc图。图9为按照实施例1的条件分析实施例7的产物得到的hplc图。具体实施方式下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。实施例1(1)仪器与色谱条件高效液相色谱仪:u3000高效液相色谱系统及工作站;蒸发光散色检测器:alltech-6000百万分之一电子天平:赛多利斯mas6.6s色谱柱:phenomenexlunac18(4.6mm×250mm,5μm)的十八烷基硅烷键合硅胶色谱柱。用三氟乙酸调节ph至2.2为水相,流动相中水相-乙腈-甲醇比例按下表1,设置流速为1.2ml/min,柱温30℃,蒸发光散色检测器:漂移管温度95℃,气体流速2.5l/min。表1(2)色谱系统适用性分别取奥贝胆酸、ob-6、ob-a、ob-b、ob-c、ob-d、ob-e、ob-f、ob-g、ob-h、ob-i对照品各适量,精密称定,用水-甲醇-乙腈=32∶18∶50溶解并稀释制成每1ml约奥贝胆酸2mg其余杂质各4.5μg的混合溶液,作为系统适用性溶液。取上述系统适用性溶液100μl,注入液相色谱仪,记录色谱图,结果见图1,可见主峰和杂质峰完全分离,分离度均大于1.5,不对称度均在0.8~1.2之间,理论塔板数均在10000以上。.实施例2(1)仪器与色谱条件高效液相色谱仪:u3000高效液相色谱系统及工作站;蒸发光散射检测器:alltech-6000百万分之一电子天平:赛多利斯mas6.6s色谱柱:phenomenexlunac18(4.6mm×250mm,5μm)的十八烷基硅烷键合硅胶色谱柱。用三氟乙酸调节ph至2.2为水相,流动相中水相-乙腈-甲醇比例按下表2,设置流速为1.2ml/min,柱温30℃,蒸发光散色检测器:漂移管温度95℃,气体流速2.5l/min。表2(2)色谱系统适用性分别取奥贝胆酸、ob-6、ob-a、ob-b、ob-c、ob-d、ob-e、ob-f、ob-g、ob-h、ob-i对照品各适量,精密称定,用水-甲醇-乙腈=36∶14∶50溶解并稀释制成每1ml约奥贝胆酸2mg其余杂质各4.5μg的混合溶液,作为系统适用性溶液。取上述系统适用性溶液100μl,注入液相色谱仪,记录色谱图。结果见图2,可见主峰和杂质峰完全分离,分离度均大于1.5,不对称度均在0.8~1.2之间,理论塔板数均在10000以上。实施例3:(1)仪器与色谱条件高效液相色谱仪:u3000高效液相色谱系统及工作站;蒸发光散色检测器:alltech-6000百万分之一电子天平:赛多利斯mas6.6s色谱柱:phenomenexlunac18(4.6mm×250mm,5μm)的十八烷基硅烷键合硅胶色谱柱。用三氟乙酸调节ph至2.2为水相,流动相中水相-乙腈-甲醇比例按下表3,设置流速为0.8ml/min,柱温30℃,蒸发光散色检测器:漂移管温度95℃,气体流速2.5l/min。表3(2)色谱系统适用性分别取奥贝胆酸、ob-6、ob-a、ob-b、ob-c、ob-d、ob-e、ob-f、ob-g、ob-h、ob-i对照品各适量,精密称定,用水-甲醇-乙腈=32∶18∶50溶解并稀释制成每1ml约奥贝胆酸2mg其余杂质各4.5μg的混合溶液,作为系统适用性溶液。取上述系统适用性溶液100μl,注入液相色谱仪,记录色谱图,结果见图3,可见主峰和杂质峰完全分离,分离度均大于1.5,不对称度均在0.8~1.2之间,理论塔板数均在10000以上。实施例4:(1)仪器与色谱条件高效液相色谱仪:u3000高效液相色谱系统及工作站;蒸发光散色检测器:alltech-6000百万分之一电子天平:赛多利斯mas6.6s色谱柱:phenomenexlunac18(4.6mm×250mm,5μm)的十八烷基硅烷键合硅胶色谱柱。用三氟乙酸调节ph至2.2为水相,流动相中水相-乙腈-甲醇比例按下表4,设置流速为1.3ml/min,柱温30℃,蒸发光散色检测器:漂移管温度95℃,气体流速2.5l/min。表4(2)色谱系统适用性分别取奥贝胆酸、ob-6、ob-a、ob-b、ob-c、ob-d、ob-e、ob-f、ob-g、ob-h、ob-i对照品各适量,精密称定,用水-甲醇-乙腈=32∶18∶50溶解并稀释制成每1ml约奥贝胆酸2mg其余杂质各4.5μg的混合溶液,作为系统适用性溶液。取上述系统适用性溶液100μl,注入液相色谱仪,记录色谱图,结果见图4,可见主峰和杂质峰完全分离,分离度均大于1.5,不对称度均在0.8~1.2之间,理论塔板数均在10000以上。实施例5:(1)仪器与色谱条件高效液相色谱仪:u3000高效液相色谱系统及工作站;蒸发光散射检测器:alltech-6000百万分之一电子天平:赛多利斯mas6.6s色谱柱:phenomenexlunac18(4.6mm×250mm,5μm)的十八烷基硅烷键合硅胶色谱柱。用三氟乙酸调节ph至2.2为水相,流动相中水相-乙腈-甲醇比例按下表5,设置流速为1.3ml/min,柱温30℃,蒸发光散色检测器:漂移管温度95℃,气体流速2.5l/min。表5(2)色谱系统适用性分别取奥贝胆酸、ob-6、ob-a、ob-b、ob-c、ob-d、ob-e、ob-f、ob-g、ob-h、ob-i对照品各适量,精密称定,用水-甲醇-乙腈=36∶14∶50溶解并稀释制成每1ml约奥贝胆酸2mg其余杂质各4.5μg的混合溶液,作为系统适用性溶液。取上述系统适用性溶液100μl,注入液相色谱仪,记录色谱图。结果见图5,可见主峰和杂质峰完全分离,分离度均大于1.5,不对称度均在0.8~1.2之间,理论塔板数均在10000以上。实施例6:(1)仪器与色谱条件高效液相色谱仪:u3000高效液相色谱系统及工作站;蒸发光散射检测器:alltech-6000百万分之一电子天平:赛多利斯mas6.6s色谱柱:phenomenexlunac18(4.6mm×250mm,5μm)的十八烷基硅烷键合硅胶色谱柱。用三氟乙酸调节ph至2.2为水相,流动相中水相-乙腈-甲醇比例按下表6,设置流速为0.8ml/min,柱温30℃,蒸发光散色检测器:漂移管温度95℃,气体流速2.5l/min。表6(2)色谱系统适用性分别取奥贝胆酸、ob-6、ob-a、ob-b、ob-c、ob-d、ob-e、ob-f、ob-g、ob-h、ob-i对照品各适量,精密称定,用水-甲醇-乙腈=36∶14∶50溶解并稀释制成每1ml约奥贝胆酸2mg其余杂质各4.5μg的混合溶液,作为系统适用性溶液。取上述系统适用性溶液100μl,注入液相色谱仪,记录色谱图。结果见图6,可见主峰和杂质峰完全分离,分离度均大于1.5,不对称度均在0.8~1.2之间,理论塔板数均在10000以上。实施例1~6的色谱图数据见表7-1、表7-2、:表7-1注:na是指其为最后一个峰,无法计算分离度。表7-2注:na是指其为最后一个峰,无法计算分离度。对比例1(1)仪器与色谱条件高效液相色谱仪:u3000高效液相色谱系统及工作站示差折光检测器:refractomax520百万分之一电子天平:赛多利斯mas6.6s色谱柱:phenomenexlunac18(4.6mm×250mm,5μm)的十八烷基硅烷键合硅胶色谱柱。用三氟乙酸调节ph至2.25为水相,流动相为水相-乙腈-甲醇=325∶145∶530,设置流速为1.2ml/min,柱温25℃,示差折光检测器:检测器温度35℃,数据采集频率10.00hz。(2)系统适用性溶液配制分别取奥贝胆酸、ob-6、ob-a、ob-b、ob-c、ob-d、ob-e、ob-f、ob-g、ob-h、ob-i对照品及其他中间体各适量,精密称定,用甲醇溶解并稀释制成每1ml约奥贝胆酸10mg其余杂质各45μg的混合溶液,作为系统适用性溶液。取上述系统适用性溶液30μl,注入液相色谱仪,记录色谱图。结果见图7,可见扩大样品浓度后ri响应还是很低,且由于ri不能用梯度洗脱,在保证杂质基线分离的情况下ob-6、ob-g不出峰,无法有效检出奥贝胆酸有关物质的含量。对比例2:(1)色谱条件:流动相:0.1%甲酸(a);乙腈(b)梯度条件:时间(分钟)a(%)b(%)07030205050409010459010色谱柱:十八烷基硅烷键合硅胶色谱柱检测器:elsd-2000检测器参数:增益:1,气流2.3l/min,漂移管温度:110℃(2)系统适用性溶液配制分别取奥贝胆酸、ob-6、ob-a、ob-b、ob-c、ob-d、ob-e、ob-f、ob-g、ob-h、ob-i对照品及其他中间体各适量,精密称定,用甲醇溶解并稀释制成每1ml约奥贝胆酸2mg其余杂质各4.5μg的混合溶液,作为系统适用性溶液。取上述系统适用性溶液100μl,注入液相色谱仪,记录色谱图。结果见图8,其中2号峰和11号峰重合,即ob-a和ob-g重合;3号峰、4号峰和5号峰重合,即ob-b,ob-f,ob-d三个峰重合;6号峰和7号峰即ob-i、ob-c,其出峰时间分别为24.98min和25.12min,分离度为1.03,故6号峰和7号峰在此条件下难以有效分离,该方法无法有效检出奥贝胆酸有关物质的含量。实施例7:奥贝胆酸原料药的生产奥贝胆酸的合成方法可按照专利或文献公开的方法合成:3α-羟基-7-酮基-5β-胆烷酸(iii)的制备向反应瓶中依次加入鹅去氧胆酸(ii)(113g,0.288mol)、溴化钠(1.78g,0.0173mol)、乙酸(30ml)和甲醇(600ml),室温搅拌至全部溶解,降温至-10℃+2℃,向反应体系中缓慢滴加13%的次氯酸钠溶液(225ml,0.39mol),控制内温在-10~0℃搅拌反应至hplc检测原料鹅去氧胆酸(ii)含量低于1%。反应完成后,撤去冰浴,反应液自然升至室温,向反应体系中滴加5%的亚硫酸氢钠溶液(25ml),搅拌30分钟,抽滤、干燥得到3α-羟基-7-酮基-5β-胆烷酸(iii)粗品(115.83g)。将粗品和甲醇(1l)加入到反应瓶中,加热至65℃,回流反应半小时,趁热过滤,将滤液重新加热回流半小时,反应液自然冷却析晶,抽滤、干燥得到白色粉末状的3α-羟基-7-酮基-5β-胆烷酸(iii)(91g,收率80.9%,hplc:99.64%)。3α-羟基-7-酮基-5β-胆烷酸甲酯(iva)的制备向三口瓶中依次加入3α-羟基-7-酮基-5β-胆烷酸(iii)(50g)、甲醇(360ml)搅拌溶解。冷却至10℃以下,滴加氯化亚砜(14ml),随后加热至65℃,回流反应3h。反应结束,冷却至5℃,向其中加入h2o(360ml),自然冷却析晶,加入晶种,加快搅拌。有大量固体析出,搅拌3小时,过滤产品,干燥得到iva(47.3g,收率91.3%),纯度98.95%。3α,7α-二-三甲基甲硅烷氧基-6-烯-5β-胆烷酸甲酯(va)的制备氮气保护下向反应瓶中加入无水四氢呋喃(260ml),干冰乙醇浴冷却至-75℃下滴加二异丙基氨基锂的四氢呋喃溶液(c=2mol/l,240ml,0.488mol),滴加完毕后搅拌10分钟,-70℃下滴加三甲基氯硅烷(112ml,0.884mol),搅拌均匀后,将3α-羟基-7-酮基-5β-胆烷酸甲酯(iva)(26.0g,0.0643mol)的四氢呋喃(100ml)溶液滴加至反应溶液中,-75℃下反应1小时。反应结束后,撤去冰浴,升温至-30℃,滴加饱和碳酸氢钠溶液(250ml),控制温度不超过20℃,静置分液,用乙酸乙酯萃取(200ml×2),合并有机相,依次用饱和碳酸氢钠(300ml×1)、饱和食盐水(500ml×1)洗涤,无水硫酸钠干燥,抽滤、减压浓缩得到黄色油状的3α,7α-二-三甲基甲硅烷氧基-6-烯-5β-胆烷酸甲酯(va)粗品(39.2g),油泵减压蒸馏30min,充氮气保护,直接用于下步反应。3α-羟基-6-亚乙基-7-酮基-5β-胆烷酸甲酯(via)的制备氮气保护下,向反应瓶中加入3α,7α-二-三甲基甲硅烷氧基-6-烯-5β-胆烷酸甲酯(va)(39.0g)和二氯甲烷(200ml),干冰/乙醇浴冷却至-60℃下,向反应瓶中加入乙醛(8.7ml),搅拌均匀后向反应液中加入三氟化硼乙醚溶液(52.5ml),滴加完毕后-60℃下反应4小时,升至室温反应3小时。反应结束后,冰水浴冷却至0-5℃下滴加饱和碳酸氢钠溶液(250ml),搅拌10分钟后,静置分液,用二氯甲烷萃取(200ml×3),合并有机相,用饱和食盐水洗涤(250ml),无水硫酸钠干燥,过滤,减压浓缩得到棕黄色油状的3α-羟基-6-亚乙基-7-酮基-5β-胆烷酸甲酯(via),油泵减压蒸馏30min,充氮气保护,直接用于下步反应。直接用于下步反应。3α-羟基-6-乙基-7-酮基-5β胆烷酸甲酯(viia)的制备将3α-羟基-6-亚乙基-7-酮基-5β-胆烷酸甲酯(via)(30g)、溶于乙醇(600ml)和盐酸(10ml)中,搅拌溶解,投入氢化斧,加入10%pd/c催化剂(0.45g),氮气置换空气三次,氢气置换氮气三次,在3atm压力下氢化反应(至氢化反应不再吸氢)。反应结束,冷却降温至40℃以下,过滤催化剂,滤液减压浓缩至干,得到类白色3α-羟基-6-乙基-7-酮基-5β胆烷酸甲酯(viia)(29.5g),收率97.8%。hplc检测构型:化学纯度96.5%,异构体比例α/β=1∶10。3α-羟基-6α-乙基-7-酮基-5β胆烷酸甲酯(viia’)的制备①向反应瓶中加入10g6-乙基-7-酮基-5β胆烷酸甲酯(viia),1.5g甲醇钠、100ml甲醇,加热回流至反应完全,用盐酸调ph为中性,加入乙酸乙醋萃取有机层,减压浓缩至干得7-酮-6α-ecdca-甲酯,转化率98.5%。②向反应瓶中加入10g6-乙基-7-酮基-5β胆烷酸甲酯(viia)、100ml氯化氢/甲醇溶液,加热回流至反应完全,减压浓缩至干得7-酮-6α-ecdca-甲酯,转化率98.9%。1hnmr(400mhz,cdcl3):δ3.67(3h,s),3.57(1h,m),2.57(1h,t,j=11.5hz),2.37(1h,m),2.24(1h,dd,j=6.6,9.6hz),2.20(1h,m),1.22(3h,s),0.93(3h,d,j=6.2hz),0.85(3h,t,j=7.3hz),0.67(3h,s).13cnmr(100mhz,cdcl3):δ215.1,178.4,71.6,56.3,51.3,50.6,50.4,47.5,46.4,44.4,43.8,40.4,38.2,36.6,36.3,35.2,32.4,32.0,30.6,29.3,25.8,23.5(2c),22.8,18.8,12.5,12.0.hrms-esim/z433.3323[m+h+]3α,7α-二羟基-6α-乙基-5β胆烷酸甲酯(viiia)的制备向反应瓶中加入3α-羟基-6-乙基-7-酮基-5β胆烷酸甲酯(viia)(24.0g)、四氢呋喃(480ml)、纯化水(60ml),搅拌溶解,冷却至10℃以下,分批次加入硼氢化钠(防止冲料),加完,于20~30℃反应1~2h。反应结束,浓缩溶剂,得到淡黄色油状物。加水(200ml)和乙酸乙酯(240ml),冰浴冷却下,用2m盐酸调节ph至3.0,水相再用乙酸乙酯(240ml)萃取,合并有机相,饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干得到类白色固体(22g)。6α-乙基-3α,7α-二羟基-5β-胆烷酸(i,奥贝胆酸)的制备将5g6α-ecdca-甲酯加入反应瓶中,加入10ml甲醇、50ml水,2eq.氢氧化钠,搅拌至反应完全。加入二氯甲烷,加入磷酸调节ph=1-2分取有机层,水洗一次,减压浓缩至干得奥贝胆酸粗品。将粗品溶于稀氨水中加入磷酸酸化析晶得奥贝胆酸精制品。收率90%,hplc纯度99.3%。运用实施例1的色谱条件,对该批次的奥贝胆酸片剂进行分析,结果见图9和表8。表8注:na是指其为最后一个峰,无法计算分离度。由图9和表8可见,该批次中奥贝胆酸的有关物质,均可以完全分离。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1