用于多糖分析的一种同位素标签试剂的制备和应用的制作方法
本发明涉及生物分子分析试剂领域,特别是用于生物分子定量分析,具体为用于多糖分析的一种同位素标签试剂的制备和应用。
背景技术:
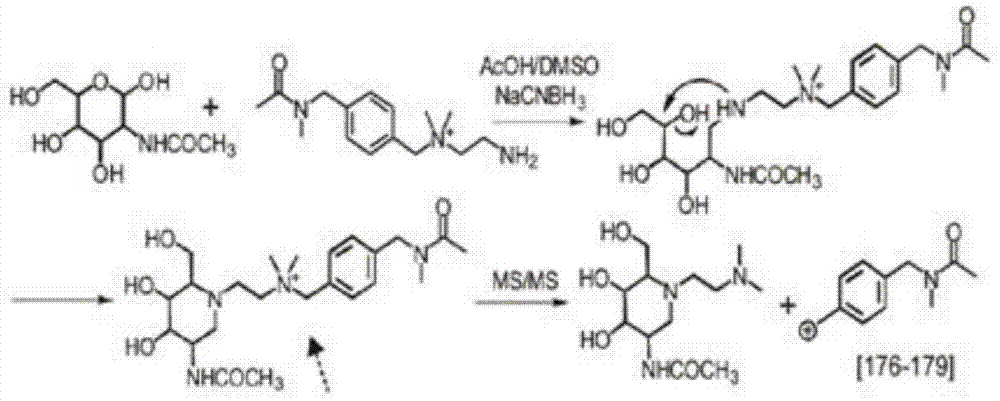
:碳水化合物是在生物生命过程中起重要作用的大分子之一,而各种糖缀合物是其它大分子的最常见修饰之一(dove,biotechnol.2001,19,913-917;dennis,j.w.etal,m.cell2009,139,1229-1241)。例如,许多蛋白质药物(例如单克隆抗体)有各种多糖修饰,其多糖的组成和化学计量可影响其分子结构的稳定性和功效(walsh,g.jefferis,r.,nat.biotechnol,2006,24,1241-1252)。因此,多糖的定性和定量测定对于生物医学和生物技术应用都具有重要意义(marino,k.nat.chem.biol.2010,6,713-723)。然而,由于与其化学性质和结构的复杂性,多糖的研究和其综合分析远远落后于其它生物分子。多糖(别称:碳水化合物或聚糖)是在生物过程如蛋白质运输,细胞与细胞间通信和免疫应答中起关键作用的一类重要的生物分子,并且它们的异常与包括癌症,痴呆和自身免疫病等许多疾病相关,治疗性生物药物如单克隆抗体的效力和稳定性也受它们携带的多糖的影响。由于这些原因,多糖分析(用于质量控制,疾病诊断等)在学术研究,制药工业和保健中具有极大的价值。然而,多糖是缺乏uv吸收并在质谱(ms)中不易电离的亲水性分子。它们的结构是高度异质的,因为每个多糖可以具有多种立体异构体。此外,多糖生物合成是非模板驱动的过程,使得它们的组成和结构难以预测。为了克服这些障碍,多糖分析以往通常被衍生化以改进它们在各种分析平台特别是质谱(ms)中的性能(ruhaak,l.r.etal.anal.bioanal.chem.2010,397,3457-3481)。用荧光2-氨基苯甲酸(2-aa)和2-氨基苯甲酰胺(2-ab)标记可以使它们与荧光检测兼容,并增强其在ms中的信号(bigge,j.c.etal.anal.biochem.1995,230,229-238)。但是由于峰共洗脱,荧光标记在定量复杂的多糖样品中是十分有限的。多糖上羟基的全甲基化可以改进它们的质谱检测,并且产生用于结构解析的高质量ms2光谱,但是实现精确的定量仍具有挑战性并且样品分析重现性较低(kang,p.etal.rapidcommun.massspectrom.2005,19,3421-3428。近年中与2-aa/2-ab标记(prien,j.m.etal.anal.chem.2010,82,1498-1508)和全甲基化(alvarez-manilla,g.etal.glycobiology2007,17,677-687)获得越来越多的关注以帮助多糖表征和定量(bowman,m.j.etal..anal.chem.2007,79,5777-5784)。存在两种类型的稳定同位素标记方法,质量移位和同量异位标签。质谱移位试剂标签使得来自不同样品的分析物在其前体质量(ms1)上产生几个道尔顿的质量差别,并且通过比较ms1相应质谱峰的强度上实现定量分析。相反,同量异位标签具有相同化学结构和分子量,但在其各种异构分子中各种重核的位置不同。因此,一旦样品被一组同量异位标签标记,它们在ms1中不显示质量差异。在ms/ms碎裂时,产生一系列不同低质量的报告离子用于定量。与质量移位标签相比,目前市售的同量异位标签,如串联质量标签(tmt)10-plex允许同时定量多达10个样品(mcalister,g.c.etal.anal.chem.2012,84,7469-7478),并且不会在ms1光谱中引入更多的复杂性,其通过来自多个样本的信号累加在一起来增加检测灵敏度。因此,其已被广泛用于大规模定量蛋白质(zhang,j.;wang,y.;li,s.anal.chem.2010,82,7588-7595;zeng,d.;li,s.chem.commun.2009,3369-3371;chen,z.etal.anal.chem.2012,84.2908-2915)和小分子代谢物(yuan,w.etal.proteomeres.2011,10,5242-5250;yuan,w.etal.anal.chem.2012,84,2892-2899)。已经有实验尝试将同量异位标记用于多糖定量(yang,s.etal.anal.chem.2013,85,8188-8195;hahne,h.etal.anal.chem.2012,84,3716-3724)。然而,由于多糖中的糖苷键在ms/ms碎裂时易于断裂并与报告离子的产生发生竞争,因此最初为多肽分析设计的同量异位标签通常不适合多糖分析。作为补救的方法,被标记的多糖必须与氯化钠(nacl)进行混合用于ms分析,这主要是因为na+可以辅助同量异位标签的片段化以产生更多的报告物离子。这种方法所引起的问题是盐离子可以严重抑制电喷雾离子源(esi)的离子化,降低esi-ms中的多糖的信号。多糖也可以形成多个h+/na+加合物,将分子复杂化,进一步降低它们的检测灵敏度。因此,多糖分析领域仍然需要新技术,特别是用于增强多糖定量分析的试剂和方法。技术实现要素:多糖作为在多个重要生物过程如蛋白质运输,细胞与细胞间通信和免疫应答中起关键作用的生物分子,它们的结构与表达量与许多复杂性疾病(例如癌症,痴呆和自身免疫病等)密切相关。同时,大多数蛋白药物(例如单克隆抗体)也携带多糖,且其在人体内的稳定性及疗效受到这些多糖的影响。因此,多糖分析有着广泛的应用。例如,本发明所提供的方法可应用于下列各种场景(但不仅限于这些场景):(1)在蛋白药物的研发与生产过程中,用于分析多糖来提高蛋白药物的疗效以及质量控制;(2)对一大类先天性糖基化病(congenitaldisorderofglycosylation)进行诊断;(3)在开发多糖疫苗(包括艾滋病等多种病毒传染病)过程中,用于研究疫苗的有效性。针对这些应用,本发明公开了n-多糖分析方法、含有季胺的同位素标签试剂及其制备方法。本发明提供如下技术方案:本发明涉及用于生物分子分析的含有季胺的同位素标签试剂的应用,以及制备和使用季胺同位素标签试剂的方法。季胺同位素标签试剂在多糖分子分析具有独特的应用,尤其是通过串联质谱法的多糖定量。本发明提供了一种n-多糖分析方法,用包含ms/ms可剪切键和能够与n-多糖缀合的反应性基团的含有季胺的同位素标签试剂标记n-多糖。作为本发明的进一步的技术方案,所述含有季胺的同位素标签试剂包括下式:报告组-平衡组-反应组,其中报告基团和平衡基团通过ms/ms可剪切键连接。作为本发明的进一步的技术方案,所述含有季胺的同位素标签试剂包括下式结构:报告基团-平衡基团-反应基团,具体结构为其中位置1-3中的至少一个包含同位素原子,报告基团和平衡基团通过ms/ms可剪切键连接,并且r是能够与多糖缀合的反应基团。作为本发明的进一步的技术方案,所述报告基团的质量在176至179da的范围内,并且所述试剂含有独立地选自13c和2h的2或3个同位素原子。作为本发明的进一步的技术方案,所述反应基团包含能够通过还原胺化与多糖缀合的反应性伯胺。本发明还提供了增强多糖的灵敏度用于糖基分析的方法,其包括用含有季胺的同位素标签试剂标记多糖。其中,所述n-多糖通过以下方法获得:(1)在固体支持物上固定包含n-多糖的获自人或非人动物血清糖蛋白;(2)化学修饰携带唾液酸的n-多糖,其化学修饰包括唾液酸的碳二亚胺偶联;(3)从固体支持物释放n-多糖。本发明中对季胺标记多糖的定量通过两步实现:(1)在ms/ms中产生特征碎片峰;(2)根据特征峰的强度对多糖进行定量。对n-多糖定量分析,所述定量分析步骤包括片段化、定量两步,标记后的n-多糖在ms/ms中产生特征碎片峰,根据特征峰的强度对n-多糖进行定量。作为本发明的进一步的技术方案,其中na+不用于定量分析标记的n-多糖。本发明还公开了一种含有季胺的同位素标签试剂(命名为qabit=quanternaryaminebasedisobarictag),含有季胺的同位素标签试剂包括下式:报告基团-平衡基团-反应基团,具体结构为其中位置1-3中的至少一个包含同位素原子,报告基团和平衡基团通过ms/ms可断裂键连接,并且r是能够与多糖缀合的反应基团。作为本发明的进一步的技术方案,r是伯胺、酰肼或氨氧基的其中一种。作为本发明的进一步的技术方案,含有季胺的同位素标签试剂为含有季胺的同量异位标签试剂。作为本发明的进一步的技术方案,其中所述报告基团的质量在176至179da的范围内,所述试剂含有独立地选自13c和2h的2或3个同位素原子,包含能够通过还原胺化与多糖缀合的反应性伯胺。作为本发明的进一步的技术方案,其中所述含有同量异序标签的季胺具有结构(i):并且其中3位的甲基是13chd2。作为本发明的进一步的技术方案,其中所述含有同量异位的标签的季胺具有结构(i):并且其中1位的碳是13c,3位的甲基是chd2。作为本发明的进一步的技术方案,其中所述含有同量异位的标签的季胺具有结构(i):并且其中2位的甲基是chd2,3位的甲基是13ch3。作为本发明的进一步的技术方案,其中所述含有同量异序标签的季胺具有结构(i):并且其中1位的碳是13c,2位的甲基是chd2。本发明还公开了一种合成制备含有季胺的同位素标签试剂方法,包括以下步骤:(a)使化合物1与化合物2反应,得到化合物3;(b)使化合物3与碳酸钠和甲基碘反应,得到化合物4;(c)使化合物4与β-巯基乙酸,氢氧化钾溶液反应,得到化合物5;(d)使化合物5与乙酸反应,得到化合物6;(e)在化合物6中使用氯化氢的乙酸乙酯溶剂除去叔丁氧羰基(boc)保护基,得到化合物7;(f)使化合物7与邻-硝基苯磺酰氯反应,得到化合物8;(g)使化合物8与碳酸钠和甲基碘反应,得到化合物9;(h)使化合物9与β-巯基乙酸,氢氧化钾溶液反应,得到化合物10;(i)使化合物10与叔丁氧羰基-2-氨基乙醛(反应物11)反应,得到化合物12;(j)使化合物12与碳酸钠和甲基碘反应,得到化合物13;(k)在化合物13中使用氯化氢的乙酸乙酯溶剂除去叔丁氧羰基(boc)保护基,得到最终产品化合物14;合成工艺如下:其中化合物1-14的iupac系统名称如下:化合物1:tert-butyl(4-(aminomethyl)benzyl)carbamate化合物2:2-nitrobenzenesulfonylchloride化合物3:tert-butyl(4-(((2-nitrophenyl)sulfonamido)methyl)benzyl)carbamate化合物4:tert-butyl(4-(((n-methyl-2-nitrophenyl)sulfonamido)methyl)benzyl)carbamate化合物5:tert-butyl(4-((methylamino)methyl)benzyl)carbamate化合物6:tert-butyl(4-((n-methylacetamido)methyl)benzyl)carbamate化合物7:(4-((n-methylacetamido)methyl)phenyl)methanaminiumchloride化合物8:n-methyl-n-(4-(((2-nitrophenyl)sulfonamido)methyl)benzyl)acetamide化合物9:n-methyl-n-(4-(((n-methyl-2-nitrophenyl)sulfonamido)methyl)benzyl)acetamide化合物10:n-methyl-n-(4-((methylamino)methyl)benzyl)acetamide化合物11:tert-butyl(2-oxoethyl)carbamate化合物12:tert-butyl(2-(methyl(4-((n-methylacetamido)methyl)benzyl)amino)ethyl)carbamate化合物13:2-((tert-butoxycarbonyl)amino)-n,n-dimethyl-n-(4-((n-methylacetamido)methyl)benzyl)ethan-1-aminium化合物14:mono(n1,n1-dimethyl-n1-(4-((n-methylacetamido)methyl)benzyl)ethane-1,2-diaminium)monochloride其中步骤(b),(d),(j)所用甲基碘,丙酸,甲基碘分别选自以下反应物对(i)-(iv)中的一组:包含同位素标记的反应物:(i)ch3i;ch3cooh;13chd2i(ii)ch3i;ch313cooh;chd2i(iii)chd2i;ch3cooh;13ch3i(iv)chd2i;13ch3cooh;ch3i。具体合成工艺为:在二氯甲烷(dcm)中混合化合物1和三乙胺,然后加入化合物2,将反应在氩气气氛下搅拌过夜,反应完成后,加入dcm,将混合物用hcl溶液洗涤两次,饱和nahco3溶液洗涤两次,并用盐水溶液洗涤一次,将有机层用无水na2so4干燥并通过rotavap除去,得到白色固体化合物3;将化合物3溶于dmf中,并加入na2co3固体。然后加入甲基碘。将反应在黑暗中搅拌2小时。通过hplc检查反应完成。反应完成后,通过rotavap除去大部分ch3i和dmf。将残余物溶于etoac和盐水溶液中,分离有机层并用60ml盐水溶液洗涤三次,用na2so4干燥并通过rotavap除去,得到微黄色固体化合物4;将化合物4溶于koh/ch3oh溶液中,加入β-巯基乙酸。将反应在氩气气氛下搅拌过夜。反应完成后,在反应混合物中有白色沉淀。过滤沉淀后,通过rotavap小心地除去甲醇。向残余物中加入水,并用etoac萃取三次,将合并的有机层用饱和nahco3溶液洗涤一次,盐水溶液=洗涤一次,将有机层用na2so4干燥并通过rotavap除去,得到浅黄色固体化合物5;将化合物5溶于dcm中,加入乙酸,然后加入edc,将反应在氩气氛围下搅拌过夜,反应完成后,先用饱和碳酸氢钠溶液洗两次,然后用盐酸溶液洗两次。溶剂干燥后,用旋转蒸发去除,得到白色固体化合物6;将化合物6溶解在2mhcl/etoac,搅拌2小时,出现大量白色沉淀,过滤并用乙酸乙酯洗涤,干燥后得到白色固体化合物7;在二氯甲烷(dcm)中混合化合物7和三乙胺,然后加入化合物2,将反应在氩气气氛下搅拌过夜,反应完成后,加入dcm,将混合物用hcl(50mmol/l)溶液洗涤两次,饱和nahco3溶液洗涤两次,并用盐水溶液洗涤一次。将有机层用无水na2so4干燥并通过rotavap除去,得到淡黄色固体化合物8;将化合物8溶于dmf中,并加入na2co3固体。然后加入甲基碘,将反应在黑暗中搅拌2小时,通过hplc检查反应完成,反应完成后,通过rotavap除去大部分ch3i和dmf,将残余物溶于etoac和盐水溶液中,分离有机层并用盐水溶液洗涤三次,用na2so4干燥并通过rotavap除去,得到微黄色固体化合物9;将化合物9溶于koh/ch3oh溶液中,加入β-巯基乙酸,将反应在氩气气氛下搅拌过夜,反应完成后,在反应混合物中有白色沉淀,过滤沉淀后,通过rotavap小心地除去甲醇。向残余物中加入水,并用etoac萃取三次,将合并的有机层用饱和nahco3溶液洗涤一次,盐水溶液洗涤一次,将有机层用na2so4干燥并通过rotavap除去,得到浅黄色固体化合物10;将化合物10溶于含有乙酸的甲醇溶液,加入克化合物11,nacnbh3溶于甲醇,慢慢加入,反应搅拌2小时后,用旋转蒸发仪除去甲醇。加入半饱和碳酸氢钠溶液,用乙酸乙酯萃取3次,合并后的有机相用饱和盐水洗后,除去有机溶剂,用二氯甲烷和乙酸乙酯作为冲洗剂,在硅胶柱层析上纯化,得到白色固体化合物12;将化合物12溶于dmf中,并加入na2co3固体,然后加入甲基碘,将反应在黑暗中搅拌2小时,通过hplc检查反应完成,反应完成后,通过rotavap除去大部分碘甲烷和dmf,所余固体在硅胶柱层析上提纯得到白色固体化合物13;将化合物13溶解在2mhcl/etoac,搅拌2小时,出现大量白色沉淀,过滤并用乙酸乙酯洗涤,干燥后得到白色固体化合物14;同位素标记的qabit的合成与未标记的化合物14的合成相同,但是从合成化合物4,化合物6,化合物13的过程中采用以下四组同位素标记反应物:(ch3i,ch3cooh,13chd2i);(ch3i,ch313cooh,chd2i);(chd2i,ch3cooh,13ch3i);(chd2i,ch313cooh,ch3i)。本发明体现新型的技术方案跟现有技术相比,所具有的优点及积极效果,它是由技术特征直接带来的必然效果。本发明具有如下有益效果:(1)由于季胺标记的多糖携带不受溶液酸碱度影响的永久性的正电荷,其在质谱中的信号大大增强,从而极大的提高了多糖分析的灵敏度,增大了罕见多糖被检测到的机会;(2)可以同时分析多个多糖样品,大大提高了多糖分析的通量,同时提高了多糖分析的重现性。在本发明的例子中,可以同时对4个样品进行分析。但是,通过简单的扩展,本发明可以对多达10个样品进行分析;(3)携带唾液酸的n-多糖通常在质谱分析中不稳定,唾液酸经常会失去,进而对多糖的分析造成误差。本发明提供的方法可以提高唾液酸在质谱中的稳定度,有助于对多糖进行精准分析。综上所述,本发明提供的多糖分析与现有方法相比,具有高灵敏度,高通量的特点,广泛适用于基础研究,药物研发,疾病诊断与治疗等场景。附图说明图1、用qabit标记的n-乙酰葡糖胺(glcnac,n-多糖还原末端上的第一残基)及其在ms2中的断裂;(箭头表示ms2中的断裂位点)图2、使用qabit对多糖进行定量分析的过程;图3、qabit试剂的合成路线;图4-图13、qabit标记n-多糖的代表性ms/ms图谱,图中箭头指出报告离子(176,177,178,179);图14-图16、qabit与市售aminoxytmt标记多糖的比较;图17-图19、qabit的定量准确度。具体实施方式下面结合附图和具体实施方式,进一步阐明本发明,应理解下述具体实施方式仅用于说明本发明而不用于限制本发明的范围。本发明涉及用于生物分子分析的含有同位素标签试剂的季胺和制备和使用含有同位素标签试剂的季胺的方法。含有同位素标签试剂的季胺特别可用于定量多糖图谱。如本文和所附权利要求书中所使用的,除非上下文另外明确指出,否则单数形式“一”,“一个”和“该”包括复数指代物。相似的数字指代相同的元件,具有结构元件的所有图都具有相同的键,即黑色正方形表示glcnac;深灰色圆圈代表甘露糖;浅灰色圆圈表示半乳糖,菱形表示唾液酸。本文中关于其特征,方面和实例各种地阐述的本发明在特定实施方式中可以被构造为包括,组成或基本上由这些特征,方面和实例中的一些或全部组成作为其元件和部件被聚合以构成本发明的各种进一步实施方式。本发明以各种排列和组合相应地设想在本发明的范围内的这些特征,方面和实例或所选择的一个或多个特征,方面和实例。本发明的试剂可用于标记或标记样品用于通过定量方法分析。本文所用的“定量”或“定量分析”是指样品组成的分析。定量允许鉴定受这种分析的样品的可测量性质,例如样品的元素的相对量,而不管样品中元素的来源。定量方法可以包括但不限于高压液相色谱(hplc),uv检测和质谱(ms)。本发明总体上涉及包含用于定量生物分子的同量异序标签的试剂。具体地,本发明涉及包含用于定量糖组学和糖组分析的同量异位标签的试剂。根据本发明的含有同量异位标签的季胺能够像糖苷键那样容易地断裂,使得标记的多糖可以产生足够的用于分析的报告物离子(图1)。特别地,这样的分析可以在不使用na+或其它金属离子的情况下进行。因此,本发明提供了含有同量异位的标签试剂的多糖反应性季胺的制备及其在定量含有同量异位标签标记的多糖的季胺中的用途,例如通过串联质谱(ms/ms)。为了提高同量异序标记的多糖的报告子离子强度,期望开发能够像糖苷键那样容易断裂的同量异位标签,使得标记的多糖可以产生强的报告子离子用于精确的多糖定量。现在已经发现,当ms/ms碎裂时,即使当其偶联到多糖上时,季胺可以容易地失去其在氮上的四个取代基之一。基于这一观察,合成了一种新型的同量异位的标签,命名为qabit:基于季胺的同量异位标签。这种同量异位的标签适用于同时对多个多糖样品的定量(图1具体描述了qabit试剂的结构和使用方法)。如本文所使用的,术语qabit可以包括根据本发明的任何基于季胺的同量异位的标签或特定结构。本发明提供了含有包含能够与多糖缀合的反应性基团的同位素标签试剂的季胺。在实施方案中,含有同位素标签试剂的季胺包含能够通过还原胺化与多糖缀合的伯胺反应性基团。根据本发明的包含同位素标签试剂的季胺通常包含报告基团-平衡基团反应性基团,其中报告基团和平衡基团通过ms/ms可剪切键连接,在ms/ms中容易断裂。在实施方案中,本发明的同量异位标签的结构包含分子量范围为系列中176至181道尔顿,优选176-179da的报告分子,补偿报告分子质量差异的平衡物质和反应性组与多糖缀合。在具体实施方案中,含有同位素标签试剂的季胺包含能够通过还原胺化与多糖缀合的伯胺反应性基团。在一些实施方案中,本发明的同位素标签试剂是含有四重季胺的试剂,其包含具有相同化学结构和分子量的一组四个分子,在不同位置具有不同的稳定核,例如13c和2h的分子。在其它实施方案中,同量异序标签试剂可以是6重或更大,在另外的位置具有重同位素原子。选择报告基团和平衡基团中的每一个中的取代基,使得归属于报告基团中的取代基的选择的质量变化被归因于平衡物中的取代基选择的质量变化所抵消组。因此,试剂的不同同位素形式将具有报告基团质量加上平衡基团质量的相同总和。在ms/ms碎裂时,用含有同位素标签试剂的季胺标记的多糖可以产生强的报告子离子用于精确的定量。此外,配合具有永久带正电荷的季胺的多糖可以增强它们在ms中的电离。因此,多糖的检测灵敏度显着增强,这在分析低丰度多糖或具有有限供应的样品时是有利的。根据本发明的包含同位素标签试剂的季胺是用于基于季胺的多糖分析和/或定量的第一同量异位标签。这些试剂使用与2-aa/2-ab类似的化学,因此可以改变用于2-aa/2-ab标记和样品制备的完善的方案。参见例如美国专利号5,747,347,其全部内容通过引用并入本文。因为季胺是永久带正电的,所以含有同量异位素标记的多糖的季胺在ms中容易电离,并且显着提高其检测限。即使同量异位标签已经广泛用于肽和小分子代谢物的定量,先前基于叔胺的糖基定量的尝试已经获得有限的成功。美国专利公开号2015/0241437,通过引用并入本文;hahne,h.“carbonyl-reactivetandemmasstagsfortheproteome-widequantificationofn-linkedglycans,anal.chem.84,3716-3724(2012).例如,市售试剂aminoxytmt标记的多糖需要形成金属离子加合物以显示足够强的报告离子,用于在断裂时定量,这可以引起ms中的离子抑制并降低检测灵敏度。这并不奇怪,因为aminoxytmt和其他基于叔胺的同量异序标签最初设计用于肽定量。由于多糖中的糖苷键比肽键更容易断裂,因此aminxytmt标记的多糖优选在多糖单元之间分开,因此产生报告子离子的效率低。发现在ms中季胺片段像糖苷键一样简单,使得开发含有同量异位标签的季胺成为可能,这使得能够同时从第一次同时实现n-多糖的全局分析。本发明的含有季胺的同位素标签具有以下通式的结构:其中a-d是有机基团。含有同量异位的标签的季胺通常具有以下结构:报告组-平衡组-反应组其中,出于多糖分析的目的,所述反应性基团能够与多糖缀合。根据本发明的含有同量异位的标签的季胺可以在季胺的氮上具有各种各样的取代基,条件是报告基团和平衡基团通过ms/ms断裂键连接。根据本发明的含有同量异位标签的季胺的同位素可以并入各种位置,并且可以是任何已知的重同位素。作为实例,季胺结构可以如下表示为4重试剂:其中重同位素可以在1-4位中的任一位置,报告基团和平衡基团通过ms/ms可断裂键连接,并且r是能够与多糖缀合的反应基团。反应基团可以是能够通过还原胺化与多糖缀合的伯胺。反应基团还可以是氨氧基,酰肼或与醛反应的其它基团。同位素可以包括15n,13c和2h。作为一个实例,这种结构可以是下式的任何结构:如下表所示(同位素以下划线显示):123176cch313chd217713cch3chd2178cchd213ch317913cchd2ch3在根据本发明的另一方面,含有季胺的同位素标签试剂具有下式:其中位置1-3中的至少一个可以包含同位素原子,报告基团和平衡基团通过ms/ms断裂键连接,并且r是反应基团并且能够与多糖缀合。反应基团可以是能够通过还原胺化与多糖缀合的伯胺。反应基团还可以是氨氧基,酰肼或与醛反应的其它基团。在本发明的一个实施方案中,提供了具有下式的含有同量异位的标签试剂的季胺报告组-平衡组-反应组其中所述试剂具有结构(i):在这样的实施方案中,报告基团和平衡基团通过ms/ms断裂键连接,报告基团具有176至179da的分子量,试剂含有2或3个独立选自13c和2h的同位素原子,2h,并且反应性基团包含能够通过还原胺化与多糖缀合的反应性伯胺。在本发明中,含有同位素标签试剂的季胺具有取代基。因此,在根据结构(i)的实施方案中,3位的甲基为13chd2。在另一个实施方案中,3位的甲基是chd2,1位的碳是13c,在另一个实施方案中,2位的甲基是chd2,3位的甲基为13ch3,在另一个实施方案中,2位的甲基是13chd2。本发明的另一方面,提供了一种n-多糖分析方法,其包括用含有包含ms/ms可断裂键的季铵标记试剂的标记n-多糖和能够与n-多糖缀合的反应性基团标记n-多糖,多糖。根据该方法的一个方面,含有同量异位的标签试剂的季胺包括下式:报告组-平衡组-反应组其中报告基团和平衡基团通过ms/ms可剪切键连接。其中重同位素可以在1-3位中的任一位置,报告基团和平衡基团通过ms/ms可断裂键连接,并且r是能够与多糖缀合的反应基团。反应基团可以是能够通过还原胺化与多糖缀合的伯胺。反应基团还可以是氨氧基,酰肼或与醛反应的其它基团。同位素可以包括13c和2h。在一些实施方案中,报告基团的质量在176至179da的范围内,并且试剂含有独立地选自13c和2h的2或3个同位素原子。在根据本发明的n-多糖分析方法的其它方面,反应性基团包含能够通过还原胺化与多糖缀合的反应性伯胺。在根据本发明的n-多糖分析方法的其它方面,所述方法还包括定量分析标记的n-多糖(图2)。在根据本发明的n-多糖分析方法的其它方面,通过以下步骤获得n-多糖(1)在固体支持物上固定包含n-多糖的糖蛋白;(2)化学修饰所述n-多糖;和(3)从固体支持物释放n-多糖。根据这些方面,蛋白质例如糖蛋白与固体支持物缀合,并且未缀合的分子可以被洗去。固定化糖蛋白上的多糖被酶促修饰或通过化学反应修饰。然后,从固体支持物释放多糖用于分析。在这些方面,固体支持物可以是已知用于这种用途的任何材料,例如聚合物珠或树脂,或可控多孔玻璃珠。在根据本发明的n-多糖分析方法的其它方面,所述糖蛋白获自生物样品,例如人或非人动物血清。如本文所用,术语“生物样品”或“生物流体”包括但不限于来自活的或以前活的受试者的任何量的物质。这样的物质包括但不限于血液,血清,血浆,尿,细胞,器官,组织,骨,骨髓,淋巴,淋巴结,滑膜组织,csf,软骨细胞,滑膜巨噬细胞,内皮细胞和皮肤。在优选的实施方案中,流体是血液或血清。在根据本发明的n-多糖分析方法的其它方面,n-多糖携带唾液酸,并且n-多糖的化学修饰包括唾液酸的碳二亚胺偶联。在根据本发明的n-多糖分析方法的其它方面,定量分析步骤包括片段化试剂和定量n-多糖。在根据本发明的n-多糖分析方法的其它方面,裂解试剂包括ms/ms可剪切键的ms/ms裂解。在根据本发明的n-多糖分析方法的其它方面,na+不用于定量分析标记的多糖。包括定量分析步骤的本发明的方法优选包括使试剂断裂和定量n-多糖,更优选使用ms/ms使试剂断裂。用于分析多糖的本发明的方法可以在不使用na+或其他金属的情况下进行。在根据本发明的方法的定量分析中不需要这样的na+,因为季胺片段在ms中像糖苷键一样容易。如图3所示,本发明另外提供了制备包含qabit试剂的季胺的方法,包括以下步骤:(a)使化合物1与化合物2反应,得到化合物3;(b)使化合物3与碳酸钠和甲基碘反应,得到化合物4;(c)使化合物4与β-巯基乙酸,氢氧化钾溶液反应,得到化合物5;(d)使化合物5与乙酸反应,得到化合物6;(e)在化合物6中使用氯化氢的乙酸乙酯溶剂除去叔丁氧羰基(boc)保护基,得到化合物7;(f)使化合物7与邻-硝基苯磺酰氯反应,得到化合物8;(g)使化合物8与碳酸钠和甲基碘反应,得到化合物9;(h)使化合物9与β-巯基乙酸,氢氧化钾溶液反应,得到化合物10;(i)使化合物10与叔丁氧羰基-2-氨基乙醛(反应物11)反应,得到化合物12;(j)使化合物12与碳酸钠和甲基碘反应,得到化合物13;(k)在化合物13中使用氯化氢的乙酸乙酯溶剂除去叔丁氧羰基(boc)保护基,得到最终产品化合物14。其中化合物1-14的iupac系统名称如下:化合物1:tert-butyl(4-(aminomethyl)benzyl)carbamate化合物2:2-nitrobenzenesulfonylchloride化合物3:tert-butyl(4-(((2-nitrophenyl)sulfonamido)methyl)benzyl)carbamate化合物4:tert-butyl(4-(((n-methyl-2-nitrophenyl)sulfonamido)methyl)benzyl)carbamate化合物5:tert-butyl(4-((methylamino)methyl)benzyl)carbamate化合物6:tert-butyl(4-((n-methylacetamido)methyl)benzyl)carbamate化合物7:(4-((n-methylacetamido)methyl)phenyl)methanaminiumchloride化合物8:n-methyl-n-(4-(((2-nitrophenyl)sulfonamido)methyl)benzyl)acetamide化合物9:n-methyl-n-(4-(((n-methyl-2-nitrophenyl)sulfonamido)methyl)benzyl)acetamide化合物10:n-methyl-n-(4-((methylamino)methyl)benzyl)acetamide化合物11:tert-butyl(2-oxoethyl)carbamate化合物12:tert-butyl(2-(methyl(4-((n-methylacetamido)methyl)benzyl)amino)ethyl)carbamate化合物13:2-((tert-butoxycarbonyl)amino)-n,n-dimethyl-n-(4-((n-methylacetamido)methyl)benzyl)ethan-1-aminium化合物14:mono(n1,n1-dimethyl-n1-(4-((n-methylacetamido)methyl)benzyl)ethane-1,2-diaminium)monochloride其中步骤(b),(d),(j)所用甲基碘,丙酸,甲基碘分别选自以下反应物对(i)-(iv)中的一组:包含同位素标记的反应物:(i)ch3i;ch3cooh;13chd2i(ii)ch3i;ch313cooh;chd2i(iii)chd2i;ch3cooh;13ch3i(iv)chd2i;13ch3cooh;ch3i。参考以下具体实施例进一步说明本发明的优点和特征,这些实施例不应被解释为以任何方式限制本发明的范围,而是在具体应用中作为本发明的一个实施方案的说明。实施例一:qabit合成如图3所述的qabit的合成路线;采用如下具体的合成路线:在30ml二氯甲烷(dcm)中混合化合物1(1.12g,4.75mmol)和1.5ml三乙胺(10.8mmol),然后加入化合物2(1g,4.52mmol)。将反应在氩气气氛下搅拌过夜。反应完成后,加入30mldcm。将混合物用50mlhcl(50mmol/l)溶液洗涤两次,50ml饱和nahco3溶液洗涤两次,并用50ml盐水溶液洗涤一次。将有机层用无水na2so4干燥并通过rotavap除去,得到1.7g白色固体化合物3(4.03mmol,89%产率)。1h-nmr(cdcl3,400mhz)δ7.60-8.00(m,4h),7.10-7.20(m,4h),4.28(s,2h),4.22(d,j=6.0hz,2h),1.45(s,9h).esi-ms(m+h)+=422.14,cal.(m+h)+=422.14。将化合物3(1.7g,4.40mmol)溶于10mldmf中,并加入2.8gna2co3(26.4mmol)固体。然后加入1.37ml甲基碘(22mmol)。将反应在黑暗中搅拌2小时。通过hplc检查反应完成。反应完成后,通过rotavap除去大部分ch3i和dmf。将残余物溶于50mletoac和50ml盐水溶液中。分离有机层并用60ml盐水溶液洗涤三次,用na2so4干燥并通过rotavap除去,得到1.8g微黄色固体化合物4(4.14mmol,94%产率)。1h-nmr(cdcl3,400mhz)δ7.60-8.00(m,4h),7.23-7.30(m,4h),4.39(s,2h),4.30(d,j=6.0hz,2h),2.76(s,3h),1.45(s,9h).esi-ms(m+h)+=436.13,cal.(m+h)+=436.14。将化合物4(2.56g,5.89mmol)溶于200ml0.5mkoh/ch3oh溶液中。加入1mlβ-巯基乙酸(14.4mmol)。将反应在氩气气氛下搅拌过夜。反应完成后,在反应混合物中有白色沉淀。过滤沉淀后,通过rotavap小心地除去甲醇。向残余物中加入120ml水,并用40mletoac萃取三次。将合并的有机层用120ml饱和nahco3溶液洗涤一次,盐水溶液120ml洗涤一次。将有机层用na2so4干燥并通过rotavap除去,得到1.0g浅黄色固体化合物5(5.4mmol,68%产率)。1h-nmr(cdcl3,400mhz)δ7.22-7.30(m,4h),4.29(d,j=6.0hz,2h),3.73(s,2h),2.44(s,3h),1.46(s,9h).esi-ms(m+h)+=251.17,cal.(m+h)+=251.18。将化合物5(1.0克,5.4mmol)溶于50mldcm中,加入乙酸(0.324克,5.4mmol),然后加入edc(1.15克,6.0mmol),将反应在氩气氛围下搅拌过夜。反应完成后,先用饱和碳酸氢钠溶液50ml洗两次,然后用50mm盐酸溶液50ml洗两次。溶剂干燥后,用旋转蒸发去除,得到1.43克白色固体化合物6(4.9mmol,90%产率)。1h-nmr(cdcl3,400mhz)δ7.22-7.30(m,4h),4.29(d,j=6.0hz,2h),3.73(s,2h),2.44(s,3h),2.20(s,3h),1.46(s,9h).esi-ms(m+h)+=293.10,cal.(m+h)+=293.18.将化合物6(1.43克,4.9mmol)溶解在50ml2mhcl/etoac,搅拌2小时,出现大量白色沉淀,过滤并用20ml乙酸乙酯洗涤,干燥后得到0.89克白色固体化合物7(3.9mmol,79%产率)。1h-nmr(cdcl3,400mhz)δ7.22-7.30(m,4h),4.29(d,j=6.0hz,2h),3.73(s,2h),2.44(s,3h),2.20(s,3h).esi-ms(m+h)+=193.50,cal.(m+h)+=193.13。在30ml二氯甲烷(dcm)中混合化合物7(0.89g,3.9mmol)和1.1ml三乙胺(7.8mmol),然后加入化合物2(0.87克,3.9mmol)。将反应在氩气气氛下搅拌过夜。反应完成后,加入30mldcm。将混合物用50mlhcl(50mmol/l)溶液洗涤两次,50ml饱和nahco3溶液洗涤两次,并用50ml盐水溶液洗涤一次。将有机层用无水na2so4干燥并通过rotavap除去,得到1.32克淡黄色固体化合物8(3.5mmol,90%产率)。1h-nmr(cdcl3,400mhz)δ7.60-8.00(m,4h),7.10-7.20(m,4h)4.29(d,j=6.0hz,2h),3.73(s,2h),2.44(s,3h),2.20(s,3h).esi-ms(m+h)+=378.45,cal.(m+h)+=378.10。将化合物8(1.3g,3.5mmol)溶于10mldmf中,并加入2.8gna2co3(26.4mmol)固体。然后加入1.37ml甲基碘(22mmol)。将反应在黑暗中搅拌2小时。通过hplc检查反应完成。反应完成后,通过rotavap除去大部分ch3i和dmf。将残余物溶于50mletoac和50ml盐水溶液中。分离有机层并用60ml盐水溶液洗涤三次,用na2so4干燥并通过rotavap除去,得到1.23克微黄色固体化合物9(3.15mmol,90%产率)。1h-nmr(cdcl3,400mhz)δ7.60-8.00(m,4h),7.10-7.20(m,4h)4.29(d,j=6.0hz,2h),3.73(s,2h),2.54(s,3h),2.44(s,3h),2.20(s,3h).esi-ms(m+h)+=392.45,cal.(m+h)+=392.12。将化合物9(1.23g,3.15mmol)溶于100ml0.5mkoh/ch3oh溶液中。加入0.5mlβ-巯基乙酸(7.2mmol)。将反应在氩气气氛下搅拌过夜。反应完成后,在反应混合物中有白色沉淀。过滤沉淀后,通过rotavap小心地除去甲醇。向残余物中加入120ml水,并用40mletoac萃取三次。将合并的有机层用60ml饱和nahco3溶液洗涤一次,盐水溶液60ml洗涤一次。将有机层用na2so4干燥并通过rotavap除去,得到1.0克浅黄色固体化合物10(2.05mmol,65%产率)1h-nmr(cdcl3,400mhz)δ7.10-7.20(m,4h)4.29(d,j=6.0hz,2h),3.73(s,2h),2.80(s,3h),2.44(s,3h),2.20(s,3h).esi-ms(m+h)+=207.64,cal.(m+h)+=207.14。将0.41克化合物10(2mmol)溶于10ml含有0.2ml乙酸的甲醇溶液,加入0.32克化合物11(2mmol),310毫克nacnbh3(5mmol)溶于10ml甲醇,慢慢加入。反应搅拌2小时后,用旋转蒸发仪除去甲醇。加入20ml半饱和碳酸氢钠溶液,用20ml乙酸乙酯萃取3次.合并后的有机相用饱和盐水洗后,除去有机溶剂,用二氯甲烷和乙酸乙酯作为冲洗剂,在硅胶柱层析上纯化,得到0.594克白色固体化合物12(1.7mmol,85%产率)1h-nmr(cdcl3,400mhz)δ7.10-7.20(m,4h)4.29(d,j=6.0hz,2h),3.73(s,2h),3.60(m,2h),3.28(m,2h),2.80(s,3h),2.44(s,3h),2.20(s,3h),1.46(s,9h).esi-ms(m+h)+=350.44,cal.(m+h)+=350.24。将化合物12(0.59g,1.7mmol)溶于10mldmf中,并加入1.4克na2co3(13.2mmol)固体。然后加入0.7ml甲基碘(12mmol)。将反应在黑暗中搅拌2小时。通过hplc检查反应完成。反应完成后,通过rotavap除去大部分碘甲烷和dmf。所余固体在硅胶柱层析上提纯得到0.44克白色固体化合物13(1.2mmol,70%产率)1h-nmr(cdcl3,400mhz)δ7.10-7.20(m,4h)4.29(d,j=6.0hz,2h),3.73(s,2h),3.60(m,2h),3.28(m,2h),2.82(s,3h),2.80(s,3h),2.44(s,3h),2.20(s,3h),1.46(s,9h).esi-ms(m+h)+=365.78,cal.(m+h)+=365.26。将化合物13(0.44克,1.2mmol)溶解在50ml2mhcl/etoac,搅拌2小时,出现大量白色沉淀,过滤并用20ml乙酸乙酯洗涤,干燥后得到0.336克白色固体化合物14(1.0mmol,85%产率)1h-nmr(cdcl3,400mhz)δ7.10-7.20(m,4h)4.29(d,j=6.0hz,2h),3.73(s,2h),3.60(m,2h),3.28(m,2h),2.82(s,3h),2.80(s,3h),2.44(s,3h),2.20(s,3h).esi-msm+=264.42,cal.m+=264.21。同位素标记的qabit的合成与未标记的化合物14的合成相同,但是从合成化合物4,化合物6,化合物13的过程中采用以下四组同位素标记反应物:(ch3i,ch3cooh,13chd2i);(ch3i,ch313cooh,chd2i);(chd2i,ch3cooh,13ch3i);(chd2i,ch313cooh,ch3i)。实施例二:血清多糖标记方法第一步,n-多糖富集。首先将20μl血清蛋白在200μl由20μl变性缓冲液(10x)和160μl缓冲液(ph10.0;40mmol/l柠檬酸钠和20mmol/l碳酸钠)组成的溶液中在100℃下变性10分钟c。在用ph10缓冲液预处理aminolinktm树脂(200μl)后,将变性蛋白质加入到300μl缓冲液(ph10.0)中的aminolinktm树脂中,并在室温下混合孵育4小时。加入50μl500mmol/l氰基硼氢化钠(1×pbs),再温育4小时。用500μl1xpbs(ph7.4)冲洗树脂两次后,用1×pbs中的50mmol/l氰基硼氢化钠(nacnbh3)还原样品4小时。将与蛋白质缀合的珠子用1mol/ltris-hcl(500μl,ph7.6)洗涤两次,剩余的醛位点用500μl1mol/ltris-hcl在50mmol/lnacnbh3(0.5小时)。珠子用400μl的1mol/lnacl洗涤三次,用h2o洗涤三次。为了稳定唾液酸残基,将固体支持物上的多糖与465μl对甲苯胺(sigma)溶液(ph4-6)孵育,其由400μl对甲苯胺,25μl36-38%hcl和40μledc(n-(3-二甲基氨基丙基)-n'-乙基碳二亚胺;5.6mol/l;sigma)。反应在室温下进行3小时,然后用400μl的1%甲酸(两次),400μl的10%乙腈(两次),400μl的1mol/lnacl(2次)两次),最后是h2o(两次)。用由4μlpngasef(newenglandbiolabs),16μl10×g7缓冲液,146μl水组成的160μl溶液释放n-多糖,在37℃孵育至少2小时。在真空干燥之前,通过carbographtm纯化洗脱的多糖。yang,s.j.;zhang,h.,anal.chem.2012,84,2232-2238。第二步:n-glvcan标记。在1mol/lnacnbh3存在下,将干燥的血清多糖重悬于80μl由二甲基亚砜(dmso)和乙酸(aa)(7:3,体积)组成的溶液混合物中。将样品以1:1:2:4(或10:10:20:40μl)的比例分成四个小瓶。将溶于dmso和aa(7:3)的混合物中的40μl100mmol/lqabit(176,177,178和179)分别加入每个样品中,并在65℃下温育4小时。通过加入2ml水和2μl浓甲酸淬灭反应。将用4重qabit标记的样品汇集用于通过carbographtm清洁。将纯化的样品溶解在400μl的0.2%甲酸中。第三步:ms/ms分析方法和参数。在与agilent6550q-tofms连接的agilent1260无限hplc-芯片立方体纳米喷雾器上进行lc-ms实验。hplc-芯片由160nl富集捕获柱和75μm×150mm分析柱(5μm,300sb)组成。流动相是:(a)具有0.1%甲酸的纳米纯水和(b)具有0.1%甲酸的乙腈。使用65分钟长的梯度方法进行lc分离。在1%b下进行在富集柱上的样品上样。用于分析柱的梯度开始于1%b,在35分钟时升至40%b,在42分钟时升至85%b,维持在85%b直至48分钟,然后在55分钟时回到1%b,并在下一次运行前平衡10分钟。以4μl/min的流速加载样品,并以0.35μl/min洗脱。q-tof在高分辨率正离子模式下操作。关键的ms参数是:源温度225℃,毛细管电压1900v,破碎电压155v,干燥气体流速13l/min。在m/z100-3000之间以ms模式下4光谱/秒的扫描速率和ms/ms模式下5光谱/秒获取数据。以斜率设置为4,偏移设置为3用于双电荷离子施加斜坡碰撞能量;斜率为2,对于三次或更高的改变离子,偏移为2。使用agilentmasshunter数据采集软件实现系统控制,并且用masshunter定性分析(b.06.00)进行数据分析。使用glycoworkbench软件(版本2.0)进行糖基鉴定。标记的n-多糖的代表性ms2光谱在图4-图13中提供。这些光谱上的大片段对于多糖结构鉴定是有用的,并且由箭头指示的报告物离子(176,177,178和179)用于多糖定量。实施例三:qabit标签的数据分析。与针对肽和小分子的其它同量异位标签一样,测试的4-重qabit试剂是具有相同化学结构和分子量的一组四个分子,但它们含有不同的稳定同位素核,如13c和2h。它们的结构由分子量范围从176至179道尔顿的系列报告分子,补偿报告分子质量差异的平衡物和通过还原胺化与多糖偶联的反应性伯胺组成。该标记化学类似于由2-aa/2-ab(2-氨基苯甲酸(2-aa)或2-氨基苯甲酰胺(2-ab)),bigge,jc等人“nonselectiveandefficientfluorescentlabeling使用2-氨基苯甲酰胺和邻氨基苯甲酸,anal.biochem.230,229-238(1995)),因此熟知的用于2-aa/2-ab标记的方案可以在没有许多改变的情况下被修改在本领域中。然而,本发明的标签和2-aa/2-ab之间的显着差异是水分子自然地和化学计量地从qabit标记的多糖中丢失,而仅2-aa/2-ab标记的多糖显示水分子的部分损失。这种现象可以通过能量上有利的六元环形成(即邻近参与反应)进行。cai,y.,ling,c.-c.&bundle,d.r."facileapproachto2-acetamido-2-deoxy-β-d-glucopyranosidesviaafuranosyloxazoline"org.lett.7,4021-4024(2005)。在ms2碎裂时,qabit标记的多糖产生强的报告离子用于精确定量,而不需要额外的正离子例如na+或金属离子,从而消除离子抑制效应并防止形成多个h+/na+加合物。此外,配合具有永久带正电荷的季胺的多糖可以增强它们在ms中的电离。因此,多糖的检测灵敏度显着增强,这在分析低丰度多糖或有限量的样品时是有利的。为了应用qabit进行多糖分析,开发了基于固相的方案,如图2所示。yang,s.,li,y.,shah,p.&zhang,h.,"glycomicanalysisusingglycoproteinimmobilizationforglycanextraction"anal.chem.85,5555-5561(2013)。糖蛋白首先固定在珠上,并在碳二亚胺偶联剂存在下用过量的对甲苯胺处理。该步骤可以在多糖上与对甲苯胺完全缀合唾液酸,以在ms分析期间稳定这些不稳定的残基。然后用pngasef从固定化的糖蛋白中释放n-多糖,导致在还原末端暴露的醛基用于含有同位素标签试剂标记的季胺。然后用反相液相色谱(rplc)偶联串联ms分析标记的多糖。这样疏水的对甲苯胺部分偶联到每个唾液酸,所以携带不同数目的唾液酸的n-多糖可以容易地解决在c18柱上。基于方程式2计算标记的多糖的单同位素质量m=fanbhcsdge。m=c+a×f+b×n+c×h+d×s+e×g+(d+e)×pt+q(2)。其中:c代表n-多糖核心结构,910.3278da;f是岩藻糖,146.0579da;n是hexnac,203.0794da;h是己糖,162.0528da;s是neu5ac,291.0954da;g是neu5gc,307.0903da;pt是对甲苯胺,89.0629da(失去一种水后),其在方案中与每个唾液酸偶联;q是qabit,233.2147da(在由于还原胺化损失一个氧并且由于相邻参与反应而损失一个水);a,b,c,d,e是每个相应单元的数目。核心结构(2hexnac和3己糖)从本文的n-多糖的式(fanbhcsdge)中排除。b和c分别表示核心结构中的hexnac和hexose单元以外的hexnac和hexose单元。在esi中,标记的多糖通常被检测为携带多重电荷(z)的离子,观察到的m/z为b.我们可以基于方程式3计算其单同位素质量(m')。方程式3:m'=b×z+(z-1.0078)为了鉴定来自ms实验的多糖,可以首先计算多糖文库中所有qabit标记的n-多糖的单同位素质量(m),例如用于功能糖组学(cfg)数据库17的联盟。然后,单同位素计算ms1光谱中离子的质量(m'),并与从多糖数据库计算的单同位素质量(m)匹配以发现其组成。实施例四:qabit标签与市销aminoxytmt用于多糖研究的比较。按照例二中说描述方法,在1%标准唾液酸糖肽(sgp)中,提取的两种n-多糖(n2h2s2>90%和n2h2s<10%)用qabit进行标记。作为比较,sgp多糖也通过市售的基于叔胺的多糖的aminoxytmt进行标记。图14显示了未标记的sgp多糖,图15显示了aminoxytmt标记的sgp多糖,图16显示了qabit标记的多糖的ms图谱。qabit标记的多糖仅显示标记为n2h2s和n2h2s2的两个单峰,表明标记反应完成。虽然aminxytmt标记的多糖也显示两个主要的峰,我们仍然观察到未标记的n2h2s和n2h2s2,表明与aminoxytmt的标记反应更难以完成。此外,aminoxytmt标记的多糖显示具有14道尔顿质量差的多个卫星峰,使ms1更复杂并平均化峰强度。该实验清楚地证明了qabit标记的完成及其超出现有市售试剂aminoxytmt的优点。实施例五:qabit定量多糖的研究。按照例二中说描述方法,通过用1:1:3:5比率的4重qabit标记来自胎牛的胎球蛋白(200μg,200μg,600μg和1000μg)多糖来测试qabit的定量准确度(图17-图19)。图17显示了qabit标记的胎球蛋白n-多糖的ms1,包括n2h2s2,n3h3s2,n3h3s3和n3h3s4。图18是代表性多糖n2h2s2的ms/ms图谱,其包括一系列多糖片段,和范围从176至179的强报告子离子。图19表明来自四个原始样品的代表性多糖n2h2s2的相对丰度。该多糖的实验结果非常接近1:1:3:5。测量的和理论的比率之间的线性相关性和来自三次独立重复的小标准偏差表明qabit定量的极好的重现性。本发明方案所公开的技术手段不仅限于上述实施方式所公开的技术手段,还包括由以上技术特征任意组合所组成的技术方案。应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也视为本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1