一种发泡塑料垫中甲酰胺含量的检测方法与流程
本发明涉及一种甲酰胺含量的检测方法,更具体地涉及一种发泡塑料垫中甲酰胺含量的检测方法,属于有机物检测技术领域。
背景技术:
甲酰胺[formamidemethanamide],分子式ch3no,又名氨基甲醛,casno.75-12-7,为透明油状液体,易挥发,具有刺激性气味。甲酰胺有两个活泼的官能团羰基和酰胺基,容易发生化学反应,生成许多含氮杂环化合物。甲酰胺能与无机酸反应,生成甲酸及铵盐。在催化剂存在下与有机卤化物或醇类反应,生成甲酸酯。还能与β-二酮、β-亚氨酮、脂肪偶联、芳烃偶联、杂环偶联反应。能与钴盐、铜盐及镍盐等结合生成络合物。甲酰胺遇五氧化二磷等强脱水剂时,可生成氰化氢。具有活泼的反应性和特殊的溶解能力,是一种用途极广的化工原料和工业溶剂。常作为软化剂、发泡剂使用,广泛应用于在农药、染料、颜料、香料、助剂领域。
瑜伽垫、儿童拼图地垫等发泡聚氨酯材质的地垫在制作过程中,经常会添加甲酰胺作为发泡剂,增加塑料产品的柔韧性使其不容易断裂。但是甲酰胺是一种中毒等级毒性化学物质,对人体健康有一定危害。急性症状以损伤神经系统为特征,容易从人的皮肤渗透到人体中,损伤血液和神经系统。由于甲酰胺具有挥发性,也可以通过呼吸道进入人体,具有致癌风险。
因此,各国对甲酰胺在瑜伽垫、儿童拼图地垫等发泡聚氨酯成分的地垫中有严格限定,禁止随意在发泡地垫用品中添加使用。根据欧盟法规条款,甲酰胺被归为1b类致癌物质。2012年6月18日,欧洲化学品管理署(echa)发布公告,正式公布第七批13项高度关注物质(svhc),其中包括甲酰胺。echa认为其“可对人类健康产生潜在的严重影响”,为致癌、致畸或具生殖毒性的物质(cmr),对眼睛和皮肤有一定的刺激,吸入或摄入后对健康也有一定的危害,长时间暴露于甲酰胺还会影响中枢神经系统。
目前国内外公开报道的关于甲酰胺检测手段主要有气相色谱法、气相色谱-质谱法,其中目前报道较多的分析方法为气相色谱-质谱联用法。现有技术的气质联用检测甲酰胺含量,主要利用乙腈进行提取,而乙腈在气相色谱串联三重四极杆方法检测中,需要溶剂转换后才能上机检测。利用气相色谱串联三重四极杆方法并直接进样检测甲酰胺含量的气相色谱串联三重四极杆方法,并且适应范围广、灵敏度高、重复性好、定性定量准确的检测方法未见报道。
因此,一种简便、快速、灵敏度高、重复性好、定性定量准确,并且能直接进样操作简单的甲酰胺含量的gc-ms-ms检测方法亟待开发出来。
技术实现要素:
本发明的目的在于克服上述现有技术的不足之处,而提供一种简便、快速、灵敏度高、重复性好、定性定量准确,并且能直接进样操作简单的发泡塑料垫中甲酰胺含量的检测方法。
为实现以上目的,本发明所采用的技术方案是:一种发泡塑料垫中甲酰胺含量的检测方法,包括如下步骤:
1)试样制备:
用洁净的工具从处于密封状态的样品上获得有代表性的试样,将样品制成5mm×5mm以下尺寸的待测样。迅速装入20ml密封试样瓶中,备用;
2)提取及净化:
准确称取0.2g(精确至0.0001g)样品,加入20ml含1%甲苯的丙酮、乙酸乙酯同体积比混合提取液,使试样全部浸入提取液中,密封振荡超声萃取。超声萃取后所得的提取液过滤至离心管中离心,离心后取上清液10ml于d-spe净化管中,充分震荡1min后静置30min,静置完成后,用含1%甲苯的丙酮、乙酸乙酯同体积比混合溶液洗涤残渣1-3次,合并洗涤液和提取液,提取液氮吹浓缩至0.5-1ml后补至1ml,将浓缩液过0.45μm滤膜,置于进样瓶中待测;
3)标准工作溶液的配制:
将不含甲酰胺的同种类基质空白样品按上述步骤1)、2)处理,得样品提取净化液,用空白提取净化液配制成至少5个浓度的甲酰胺系列混合标准工作液;
4)测定和结果计算:
将步骤3)中的各浓度梯度的标准工作溶液进行gc-ms-ms测定,以标准工作溶液的色谱峰面积对其相应浓度进行回归分析,得到基质标准工作曲线;在相同条件下将步骤2)中净化后的样品液进行gc-ms-ms测定,测得样品液中甲酰胺的色谱峰面积,代入基质标准曲线,得到样品液中甲酰胺含量,然后根据样品液所代表试样的质量计算得到样品中甲酰胺含量。
优选的,所述的步骤2)中超声萃取的温度为50-80℃,提取时间0.5-1.5h。
优选的,所述的步骤2)中提取液离心条件为4000r/min离心5min。
优选的,所述的步骤2)中的d-spe净化管为15mltube,900mgmgso4/450mgpsa/300mgc18/50mggcb。
优选的,所述的步骤3)中配制成1.0、2.0、5.0、10.0、25.0、50.0μg/ml基质标准工作溶液浓度的甲酰胺系列混合标准工作溶液。
测得样品溶液中未知组分的保留时间(rt)分别与标准溶液在同一色谱柱上的保留时间(rt)相比较,如果样品溶液中某组分的保留时间与标准溶液保留时间相差在±0.05min内,并且该峰同时具有定量离子对和定性离子对的可认定为该成分。若上述两个条件不能同时满足,则判断不含该种成分。
色谱条件:色谱柱:tr-5ms毛细管色谱柱,柱长30m,内径0.25mm,膜厚0.25μm;进样口温度240℃;载气:氦气(≥99.999%);不分流模式进样,进样量:1μl;恒流模式,流速1.0ml/min;升温程序:初温70℃,保持2min,以每分钟10℃的速度升至150℃,然后以每分钟30℃的速度升至230℃,保持2min;传输线温度:230℃。
电离模式:ei模式,能量70ev;离子源温度230℃;扫描方式:二级离子监测(srm)模式;定量离子对为:17、29、44、45,碰撞电压25ev,定性离子对为:45,碰撞电压20ev。
试样中被测甲酰胺含量以质量分数ω计,单位以微克每毫升(μg/ml)表示,按下式计算。
ρ——标准溶液中甲酰胺的质量浓度,单位为微克每毫升(μg/ml);
a——样品溶液中甲酰胺的峰面积;
as——标准溶液中被测甲酰胺的峰面积;
v——提取溶剂总体积,单位为毫升(ml);
m——试样的质量,单位为克(g)。
以标准工作溶液的色谱峰面积对其相应浓度进行回归分析,得到标准工作曲线。
本发明所具有的有益效果:
1、本发明采用改样品前处理提取和净化步骤使用含有1%甲苯的丙酮、乙酸乙酯同体积比混合溶液代替乙腈作为提取液,直接提取净化后去上清液直接进样而不需要溶剂转换,克服了乙腈不能直接进gc-ms-ms的缺陷。
2、本发明采用气相色谱-三重四级杆串联质谱(gc-ms-ms)定性定量测定发泡塑料垫中甲酰胺的含量,平均回收率为87.2%~96.3%,平均相对标准偏差(rsd)为1.11%~3.2%,灵敏度高、重复性好。
附图说明
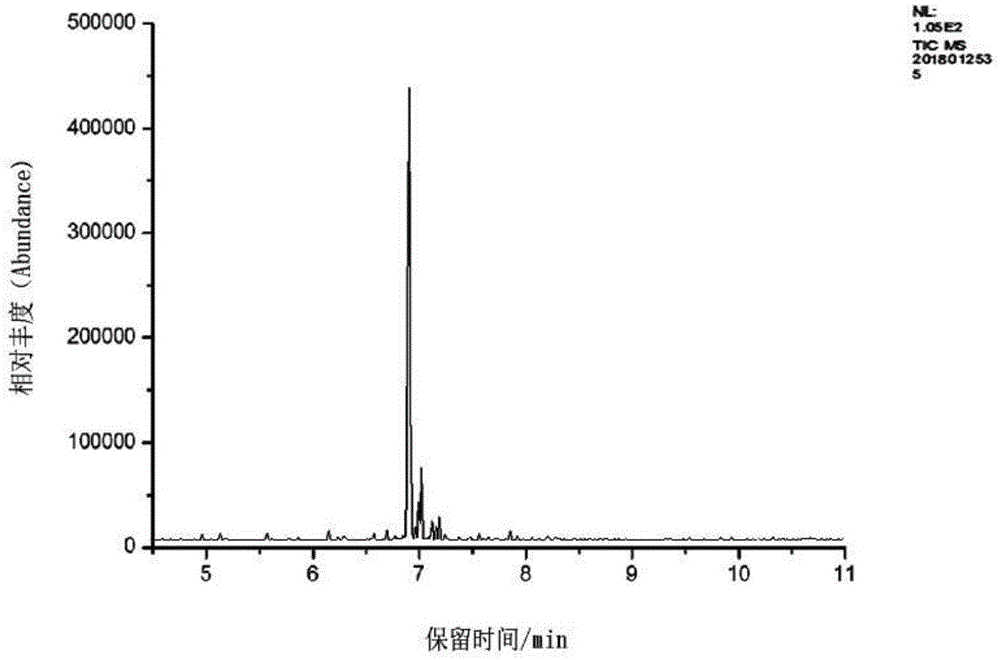
图1甲酰胺离子流色谱图;
图2甲酰胺质谱图;
图3甲酰胺质谱峰面积/质量浓度标准曲线。
具体实施例
下列实施例是对发明的进一步说明,但本发明不仅限于此。
实施例中使用的仪器:
trace1310型气相色谱-tsq8000型三重四极杆串联质谱仪(美国thermo公司);sartorius电子天平(千分之一,北京赛多利斯天平有限公司);ikat18basic均质器(德国ika公司);ms3基本型涡旋混合器(ika,germany)。
试剂标准品:
标准品购自天津农业部环境质量监督检验测试中心(质量浓度均为100ug/ml)。丙酮、甲苯(分析纯,国药集团化学试剂有限公司),乙酸乙酯(分析纯,天津市广成化学试剂有限公司),乙酸钠、氯化钠(分析纯,天津博迪化工股份有限公司)。
色谱条件:色谱柱:tr-5ms毛细管色谱柱,柱长30m,内径0.25mm,膜厚0.25μm;进样口温度240℃;载气:氦气(≥99.999%);不分流模式进样,进样量:1μl;恒流模式,流速1.0ml/min;升温程序:初温70℃,保持2min,以每分钟10℃的速度升至150℃,然后以每分钟30℃的速度升至230℃,保持2min;传输线温度:230℃。
电离模式:ei模式,能量70ev;离子源温度230℃;扫描方式:二级离子监测(srm)模式;定量离子对为:17、29、44、45,碰撞电压25ev,定性离子对为:45,碰撞电压20ev。
实施例1瑜伽垫中甲酰胺含量的gc-ms-ms检测:
1)试样制备:
用洁净的工具从处于密封状态的瑜伽垫样品上获得有代表性的试样,将样品制成5mm×5mm以下尺寸的待测样。迅速装入20ml密封试样瓶中,备用;
2)提取及净化:
准确称取0.2g(精确至0.0001g)样品,加入20ml含1%甲苯的丙酮、乙酸乙酯同体积比混合提取液,使试样全部浸入提取液中,50℃密封振荡超声萃取0.5-1.5h。超声萃取后所得的提取液过滤至离心管中,4000r/min离心5min,离心后取上清液10ml于d-spe(15mltube,900mgmgso4/450mgpsa/300mgc18/50mggcb)净化管中,充分震荡1min后静置30min,静置完成后,用含1%甲苯的丙酮、乙酸乙酯同体积比混合溶液洗涤残渣1次,合并洗涤液和提取液,提取液氮吹浓缩至0.5ml后补至1ml,将浓缩液过0.45μm滤膜,置于进样瓶中待测;
3)标准工作溶液的配制:
将不含甲酰胺的同种类基质空白样品按上述步骤1)、2)处理,得样品提取净化液,用空白提取净化液配制成1.0、2.0、5.0、10.0、25.0、50.0μg/ml浓度的甲酰胺系列混合标准工作液;
4)测定和结果计算:
将步骤3)中的各浓度梯度的标准工作溶液进行gc-ms-ms测定,以标准工作溶液的色谱峰面积对其相应浓度进行回归分析,得到基质标准工作曲线;在相同条件下将步骤2)中净化后的样品液进行gc-ms-ms测定,测得样品液中甲酰胺的色谱峰面积,代入基质标准曲线,得到样品液中甲酰胺含量,然后根据样品液所代表试样的质量计算得到样品中甲酰胺含量。
试样中被测甲酰胺含量以质量分数ω计,单位以微克每毫升(μg/ml)表示,按下式计算。
ρ——标准溶液中甲酰胺的质量浓度,单位为微克每毫升(μg/ml);
a——样品溶液中甲酰胺的峰面积;
as——标准溶液中被测甲酰胺的峰面积;
v——提取溶剂总体积,单位为毫升(ml);
m——试样的质量,单位为克(g)。
线性回归方程:甲酰胺y=42286x+1913.75,相关系数0.9999。
加标回收率和重复性:在不含甲酰胺的瑜伽垫中加入1.0、5.0和25.0μg/ml3个浓度水平的甲酰胺标准溶液,添加30min后按上述处理步骤进行含量测定,0.2mg/kg添加浓度的样品待测液用测定前,用1%甲苯的丙酮、乙酸乙酯同体积比混合溶剂将净化定容液稀释至甲酰胺检出量在线性范围之内。将测定浓度与甲酰胺理论添加浓度进行比较,得到甲酰胺添加回收率,每个添加水平平行测定6次,得其相对标准偏差,测定结果见表1。由表1可以看出,在3个加标水平上,甲酰胺的平均回收率为89.2%~96.3%,平均相对标准偏差(rsd)为1.58%~3.23%,说明本发明方法的回收率较高,重复性好。
表1实施例1中甲酰胺的回收率和重复性(n=6)
实施例2儿童拼图坐垫中甲酰胺含量的gc-ms-ms检测:
1)试样制备:
用洁净的工具从处于密封状态的瑜伽垫样品上获得有代表性的试样,将样品制成5mm×5mm以下尺寸的待测样。迅速装入20ml密封试样瓶中,备用;
2)提取及净化:
准确称取0.2g(精确至0.0001g)样品,加入20ml含1%甲苯的丙酮、乙酸乙酯同体积比混合提取液,使试样全部浸入提取液中,65℃密封振荡超声萃取1h。超声萃取后所得的提取液过滤至离心管中,4000r/min离心5min,离心后取上清液10ml于d-spe(15mltube,900mgmgso4/450mgpsa/300mgc18/50mggcb)净化管中,充分震荡1min后静置30min,静置完成后,用含1%甲苯的丙酮、乙酸乙酯同体积比混合溶液洗涤残渣2次,合并洗涤液和提取液,提取液氮吹浓缩至1ml后补至1ml,将浓缩液过0.45μm滤膜,置于进样瓶中待测;
3)标准工作溶液的配制:
将不含甲酰胺的同种类基质空白样品按上述步骤1)、2)处理,得样品提取净化液,用空白提取净化液配制成1.0、2.0、5.0、10.0、25.0、50.0μg/ml浓度的甲酰胺系列混合标准工作液;
4)测定和结果计算:
将步骤3)中的各浓度梯度的标准工作溶液进行gc-ms-ms测定,以标准工作溶液的色谱峰面积对其相应浓度进行回归分析,得到基质标准工作曲线;在相同条件下将步骤2)中净化后的样品液进行gc-ms-ms测定,测得样品液中甲酰胺的色谱峰面积,代入基质标准曲线,得到样品液中甲酰胺含量,然后根据样品液所代表试样的质量计算得到样品中甲酰胺含量。
试样中被测甲酰胺含量以质量分数ω计,单位以微克每毫升(μg/ml)表示,按下式计算。
ρ——标准溶液中甲酰胺的质量浓度,单位为微克每毫升(μg/ml);
a——样品溶液中甲酰胺的峰面积;
as——标准溶液中被测甲酰胺的峰面积;
v——提取溶剂总体积,单位为毫升(ml);
m——试样的质量,单位为克(g)。
线性回归方程:甲酰胺y=42286x+1824,相关系数0.9999。
加标回收率和重复性:在不含甲酰胺的瑜伽垫中加入2.0、10.0和50.0μg/ml3个浓度水平的甲酰胺标准溶液,添加30min后按上述处理步骤进行含量测定,0.2mg/kg添加浓度的样品待测液用测定前,用1%甲苯的丙酮、乙酸乙酯同体积比混合溶剂将净化定容液稀释至甲酰胺检出量在线性范围之内。将测定浓度与甲酰胺理论添加浓度进行比较,得到甲酰胺添加回收率,每个添加水平平行测定6次,得其相对标准偏差,测定结果见表2。由表2可以看出,在3个加标水平上,甲酰胺的平均回收率为87.2%~92.3%,平均相对标准偏差(rsd)为1.50%~2.24%,说明本发明方法的回收率较高,重复性好。
表2实施例2中甲酰胺的回收率和重复性(n=6)
实施例3瑜伽垫中甲酰胺含量的gc-ms-ms检测:
1)试样制备:
用洁净的工具从处于密封状态的瑜伽垫样品上获得有代表性的试样,将样品制成5mm×5mm以下尺寸的待测样。迅速装入20ml密封试样瓶中,备用;
2)提取及净化:
准确称取0.2g(精确至0.0001g)样品,加入20ml含1%甲苯的丙酮、乙酸乙酯同体积比混合提取液,使试样全部浸入提取液中,80℃密封振荡超声萃取1.5h。超声萃取后所得的提取液过滤至离心管中,4000r/min离心5min,离心后取上清液10ml于d-spe(15mltube,900mgmgso4/450mgpsa/300mgc18/50mggcb)净化管中,充分震荡1min后静置30min,静置完成后,用含1%甲苯的丙酮、乙酸乙酯同体积比混合溶液洗涤残渣3次,合并洗涤液和提取液,提取液氮吹浓缩至1ml后补至1ml,将浓缩液过0.45μm滤膜,置于进样瓶中待测;
3)标准工作溶液的配制:
将不含甲酰胺的同种类基质空白样品按上述步骤1)、2)处理,得样品提取净化液,用空白提取净化液配制成1.0、2.0、5.0、10.0、25.0、50.0μg/ml浓度的甲酰胺系列混合标准工作液;
4)测定和结果计算:
将步骤3)中的各浓度梯度的标准工作溶液进行gc-ms-ms测定,以标准工作溶液的色谱峰面积对其相应浓度进行回归分析,得到基质标准工作曲线;在相同条件下将步骤2)中净化后的样品液进行gc-ms-ms测定,测得样品液中甲酰胺的色谱峰面积,代入基质标准曲线,得到样品液中甲酰胺含量,然后根据样品液所代表试样的质量计算得到样品中甲酰胺含量。
试样中被测甲酰胺含量以质量分数ω计,单位以微克每毫升(μg/ml)表示,按下式计算。
ρ——标准溶液中甲酰胺的质量浓度,单位为微克每毫升(μg/ml);
a——样品溶液中甲酰胺的峰面积;
as——标准溶液中被测甲酰胺的峰面积;
v——提取溶剂总体积,单位为毫升(ml);
m——试样的质量,单位为克(g)。
线性回归方程:甲酰胺y=42286x-268.2,相关系数0.9999。
加标回收率和重复性:在不含甲酰胺的瑜伽垫中加入1.0、10.0和50.0μg/ml3个浓度水平的甲酰胺标准溶液,添加30min后按上述处理步骤进行含量测定,0.2mg/kg添加浓度的样品待测液用测定前,用1%甲苯的丙酮、乙酸乙酯同体积比混合溶剂将净化定容液稀释至甲酰胺检出量在线性范围之内。将测定浓度与甲酰胺理论添加浓度进行比较,得到甲酰胺添加回收率,每个添加水平平行测定6次,得其相对标准偏差,测定结果见表3。由表3可以看出,在3个加标水平上,甲酰胺的平均回收率为90.9%~94.6%,平均相对标准偏差(rsd)为1.11%~1.54%,说明本发明方法的回收率较高,重复性好。
表3实施例3中甲酰胺的回收率和重复性(n=6)
数据表明,将实施例方法配制的1.0、2.0、5.0、10.0、25.0、50.0μg/ml六种浓度基质匹配标准溶液以待测物定量离子的峰面积(y)对质量浓度(x,μg/ml)做标准曲线,得到甲酰胺在三种发泡塑料垫中含量的线性方程及相关系数。结果显示,甲酰胺1.0~50.0μg/ml范围内线性关系良好,其相关系数均大于0.98。在空白基质中添加1.0、2.0、5.0μg/ml三个浓度的标准溶液,按照实验方法进行提取、净化、上机检测,方法的灵敏度较高。
上述实施例仅是对本发明的优选实施方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通工程技术对本发明的技术方案作出的各种变型和改进,均应落入本发明的权利要求书确定的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!