一种尿液中苯丙胺类物质的SPME-GC检测方法与流程
本发明涉及一种尿液中苯丙胺类物质的spme-gc检测方法,属于分析化学领域。
背景技术:
苯丙胺类毒品是一类人工合成的有机胺类兴奋剂,具有强烈兴奋作用,属于违禁毒品。苯丙胺的滥用可通过对血液、尿液、唾液及毛发等生物检材中的毒品进行检测加以证实。由于体内毒品的浓度会随着时间代谢而逐渐降低,因此,建立灵敏、准确且简单快速的检测方法用于测定生物样品中痕量毒品非常必要。
根据国内外研究成果,苯丙胺类毒品检测方法主要包括气相色谱法、高效液相色谱法、毛细管电泳法、高效液相色谱法-质谱联合检测,但通常在测试前需要进行复杂的前处理,包括溶剂萃取、浓缩等,操作繁琐,效率低下。本发明采用多孔聚三苯胺/凝胶混合涂层应用到固相萃取技术,建立了简单快速的前处理方法,联合气相色谱检测,灵敏度高,选择性强,为苯丙胺的检测提供更高效简捷的方法。固相微萃取(solid-phasemicroextraction,spme)技术是集采样、萃取、浓缩和进样于一体的样品前处理技术,已被广泛应用到环境工程,生物工程,医药工程以及食品工程中。将spme技术应用于吸毒者尿液中苯丙胺的检测将有效提高灵敏度和效率。
spme技术的核心在于萃取纤维(或称萃取头),而萃取纤维的品质取决于萃取涂层对分析物的富集效率、选择性及其稳定性。根据萃取原理不同,可分为吸收涂层和吸附涂层,其中后者具有更高的灵敏度和选择性,跟适于痕量物质的检测。吸附涂层的功能组分是固态多孔材料,多孔有机骨架材料(porousorganicframeworks,pofs)是一种具有规则孔道的新型固态多孔材料,由于其结构的多样性及可调控性吸引了人们的注意。因此研究出合适的pofs材料用于检测尿液中苯丙胺类物质具有重要意义。
技术实现要素:
本发明旨在提供一种操作简单,灵敏度高,准确性高的spme联合气相色谱法(gc),用于吸毒者尿液中的苯丙胺类物质的检测。
为解决上述技术问题,本发明采用的技术方案为:
一种尿液中苯丙胺类物质的spme-gc检测方法,包括以下步骤:
(1)苯丙胺和甲基苯丙胺用水稀释至所需浓度作为标准工作溶液;
(2)取标准工作溶液,浓度由低至高依次测试,置于顶空瓶中,用naoh将溶液ph值调至14,加入1.5-3mgnacl,搅拌均匀,将顶空瓶封口;
(3)将paf-41涂层纤维连接到5μl的微量进样针上,作为固相微萃取纤维;
(4)将(3)中提到的固相微萃取纤维插入顶空瓶,推出固相微萃取纤维头,置于样品溶液顶空位置,在40℃条件下,保持20-60min,搅拌速度为400-600r/min;
(5)将萃取纤维头抽回微量进样器中,随后将微量进样器拔出,立刻插入气相色谱进样口,再次推出萃取头,进样口温度为200-250℃,解释时间为0.5-3min;
(6)经气相色谱分离,离子火焰检(fid)测器检测,将测得的峰面积(y)与标准溶液的质量浓度(x)进行线性回归分析,得到各目标化合物的标准曲线方程;
(7)取含有苯丙胺的尿液样品1ml,用水稀释,按照上述1-6步骤中标准工作溶液的测试方法,对尿液样品进行测定,以保留时间定性,以测得目标物峰面积值,代入标准曲线方程,求得样品中的苯丙胺和甲基苯丙胺的含量。
优选地:所述的苯丙胺和甲基苯丙胺标准溶液的浓度为10,50,100,500μg/l。
优选地:所述的步骤2中标准工作溶液的体积为5-10ml。
优选地:所述的步骤7中1ml尿液样品稀释至5-10ml。
优选地:所述的色谱条件为:db-5msui型毛细管柱(30m×0.25mmi.d.×0.25μm);载气he流速为1.0ml/min,碰撞气n2流量为40ml/min,合成气(n2:o2=79:21)流速为40ml/min,氢气流速为400ml/min;,检测器温度250℃,无分离模式2分钟;升温程序:70℃起始温度以10℃/min速度升至250℃保持10min。
优选地:所述的paf-41涂层纤维的制备方法,包括以下步骤:
(1)再将表面羟基化的基底纤维一端插入该混合物到辅助胶中0.5-30分钟,缓慢后取出,再插入到制备的paf-41粉末中,然后50-90℃干燥2-60分钟,重复上述操作,直到达到所需的涂层厚度;
(2)最后在200-250℃条件下老化1-5小时,得到spme萃取纤维。
优选地:所述辅助胶为聚二甲基硅氧烷,聚酰亚胺的预聚物或者环氧乙烷胶黏剂。
优选地:所述的涂层厚度为20-100μm。
优选地:所述的paf-41的制备方法:
(1)在氮气保护下,向反应器中加入等摩尔比的双-(1,5-环辛二烯)镍和2,2-联吡啶,随后注入体积比为1:10-1:100的无水1,5-环辛二烯和无水n,n-二甲基乙酰胺,其中双-(1,5-环辛二烯)镍的浓度为0.01-0.5mol·l-1,将混合物在80-95℃条件下加热搅拌0.5-5小时,以活化催化剂;
(2)同样,在氮气保护下,将三(4-溴苯)胺溶解于无水n,n-二甲基乙酰胺中,浓度为0.01-0.1mol·l-1,80-95℃条件下搅拌0.5-5小时;
(3)随后,将上述两溶液混合,整个反应体系在80-95℃下加热搅拌72-120小时;
(4)待反应体系的温度降到室温之后,加入浓盐酸至颜色变为翠绿色过滤得到粗产物;
(5)并用大量蒸馏水洗至溶液呈中性,再用乙醇和氯仿清洗粗产品3-10次,以除去未充分反应的原料和催化剂,最后,在80-200℃条件下抽真空,得到paf-41。
优选地:所述的paf-41的另一种制备方法:
(1)在氮气保护下,将无水路易斯酸催化剂加入反应器中,注入无水chcl3(催化剂的浓度为0.02-0.2mol·l-1),60-90℃搅拌3小时;
(2)1,3,5-三苯胺溶于无水chcl3(浓度为0.02-0.2mol·l-1)后;
(3)将步骤(2)溶液注入到步骤(1)体系中,催化剂与1,3,5-三苯胺的mol比为0.4-3:5,60-90℃反应1-5天;
(4)反应结束,且温度降至室温后,过滤,分别用盐酸(1-5mol·l-1),甲醇,丙酮洗涤产物,随后依次用乙醇,四氢呋喃,氯仿对产物进行索式提取,提取时间为1-3天;
(5)最后,在80-200℃条件下抽真空,得到paf-41。
优选地:所述的路易斯催化剂为alcl3,fecl3,cucl2
优选地:所述的表面羟基化的基底纤维的制备方法为:将基底纤维的一端浸泡到氢氧化钠溶液中,然后,用盐酸溶液中和多余的氢氧根离子,最后,纤维用去离子水清洗在空气中干燥。
本发明与现有技术相比具有以下优点:
(1)本发明所使用的spme纤维是paf-41涂层纤维,paf-41是一种新型的多孔芳香骨架材料,与商品化以及已报道的纤维涂层相比,它的结构特点使其对苯丙胺和甲基苯丙胺具有特有的选择性吸附性能。这是因为:第一,具有较大的比表面积,均匀的孔径分布,对挥发性有机物具有很好的吸附性能;第二,该材料具有很高的芳香性,与目标分析物苯丙胺和甲基苯丙胺之间极易形成π-π相互作用,有利于目标物的萃取;第三,该多孔材料的骨架中很有n原子,通过相似相容原理、氢键等可大大增强其对目标分析无中-nh2的吸引,从而提高萃取能力。
(2)本发明涉及的方法不需要溶剂萃取等前处理步骤,也不需要衍生步骤,方便,高效,快捷,处理时间小于60min。
附图说明
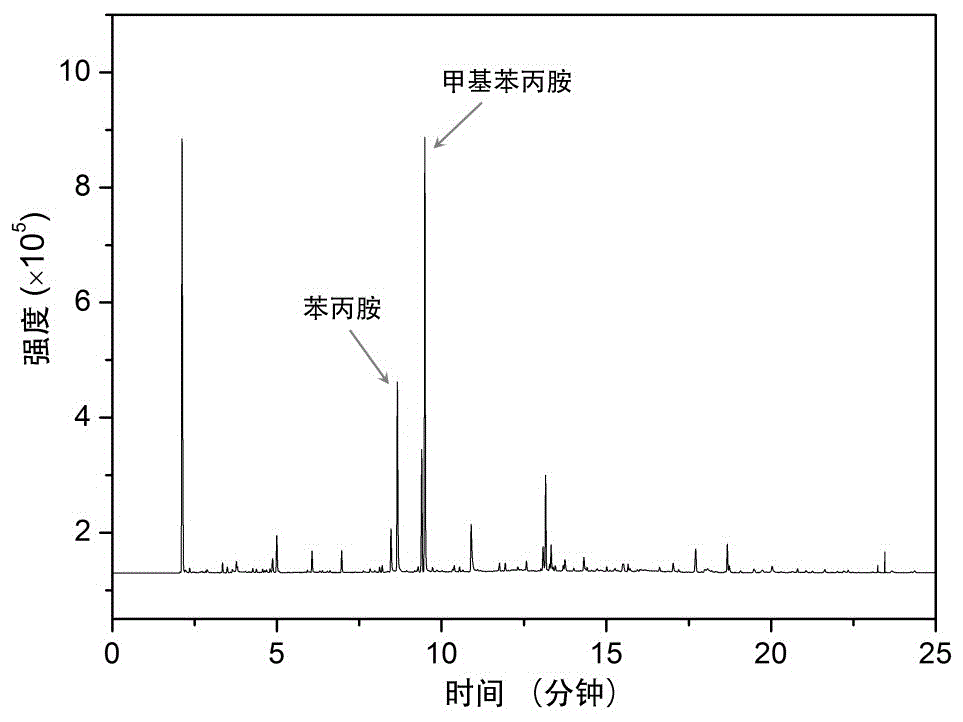
图1本发明实施例1中的色谱图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
一种paf-41涂层纤维的制备方法,包括以下步骤:
(1)在氮气保护下,向反应器中加入等摩尔比的双-(1,5-环辛二烯)镍和2,2-联吡啶,随后注入无水1,5-环辛二烯和无水n,n-二甲基乙酰胺(体积比为1:50),其中双-(1,5-环辛二烯)镍的浓度为0.16mol·l-1。将混合物在80℃条件下搅拌1小时,以活化催化剂。
(2)在氮气保护下,将三(4-溴苯)胺溶解于无水n,n-二甲基乙酰胺中,浓度为0.014mol·l-1。80℃条件下搅拌1小时。
(3)将步骤2溶液注入到步骤1体系中,并在80℃下搅拌反应72小时。
(4)反应结束,且体系温度降到室温之后,加入浓盐酸(12mol·l-1)至颜色变为翠绿色过滤得到粗产物。
(5)用大量蒸馏水洗至溶液呈中性、再依次分别用乙醇和氯仿清洗粗产品5次,以除去未充分反应的原料和催化剂。最后,在80℃条件抽真空12小时下,得到paf-41。
(6)将甲基三甲氧基硅烷300μl,羟基端头的聚二甲基硅氧烷180mg和聚甲基氢硅氧烷30mg加入离心管中,涡旋震荡5分钟。随后加入150μl三氟乙酸(95%),再次涡旋震荡5分钟。
(7)将不锈钢纤维(直径0.1mm,长6cm)的一端(3cm)浸泡到浓度为0.5-2mol.l-1的氢氧化钠溶液中,然后,用浓度为0.5-2mol.l-1的盐酸溶液中和多余的氢氧根离子,最后,纤维用去离子水清洗在空气中干燥,得到表面羟基化的不锈钢纤维;
(8)将表面羟基化的不锈钢纤维的羟基化的一端插入步骤6得到的混合物中5分钟后,缓慢取出,再插入步骤5得到的paf-41粉末中,然后60℃干燥2min;重复3次,直到达到所需的涂层厚度约60μm.
(9)将步骤8得到的纤维置于240℃条件下老化2小时,得到paf-41涂层纤维。
实施例2
一种paf-41涂层纤维的制备方法,包括以下步骤:
(1)在氮气保护下,向反应器中加入等摩尔比的双-(1,5-环辛二烯)镍和2,2-联吡啶,随后注入无水1,5-环辛二烯和无水n,n-二甲基乙酰胺(体积比为1:10),其中双-(1,5-环辛二烯)镍的浓度为0.01mol·l-1。将混合物在90℃条件下搅拌0.5小时,以活化催化剂。
(2)在氮气保护下,将三(4-溴苯)胺溶解于无水n,n-二甲基乙酰胺中,浓度为0.01mol·l-1,90℃条件下搅拌0.5小时。
(3)将步骤2溶液注入到步骤1体系中,并在90℃下搅拌反应100小时。
(4)反应结束,且体系温度降到室温之后,加入浓盐酸(12mol·l-1)至颜色变为翠绿色过滤得到粗产物。
(5)用大量蒸馏水洗至溶液呈中性、再依次分别用乙醇和氯仿清洗粗产品3次,以除去未充分反应的原料和催化剂,最后,在120℃条件抽真空10小时下,得到paf-41。
(6)将石英纤维(直径0.1mm,长6cm)的一端(1cm)浸泡到浓度为1mol.l-1的氢氧化钠溶液中,然后,用浓度为1mol.l-1的盐酸溶液中和多余的氢氧根离子,最后,纤维用去离子水清洗在空气中干燥,得到表面羟基化的石英纤维;
(7)将甲基三甲氧基硅烷300μl,羟基端头的聚二甲基硅氧烷180mg和聚甲基氢硅氧烷30mg加入离心管中,涡旋震荡5分钟。随后加入150μl三氟乙酸(95%),再次涡旋震荡5分钟。
(8)将表面羟基化的石英纤维的羟基化的一端插入步骤7得到的混合物中0.5分钟后,缓慢取出,再插入到步骤5得到的paf-41的粉末中,50℃干燥2min;重复2次,直到达到所需的涂层厚度约20μm.
(9)将步骤8得到的纤维置于200℃条件下老化5小时,得到paf-41涂层纤维。
实施例3
一种paf-41涂层纤维的制备方法,包括以下步骤:
(1)在氮气保护下,向反应器中加入等摩尔比的双-(1,5-环辛二烯)镍和2,2-联吡啶,随后注入无水1,5-环辛二烯和无水n,n-二甲基乙酰胺(体积比为1:50),其中双-(1,5-环辛二烯)镍的浓度为0.5mol·l-1。将混合物在95℃条件下搅拌5小时,以活化催化剂。
(2)在氮气保护下,将三(4-溴苯)胺溶解于无水n,n-二甲基乙酰胺中,浓度为0.1mol·l-1,95℃条件下搅拌0.5小时。
(3)将步骤2溶液注入到步骤1体系中,并95℃下搅拌反应120小时。
(4)反应结束,且体系温度降到室温之后,加入浓盐酸(12mol·l-1)至颜色变为翠绿色过滤得到粗产物。
(5)用大量蒸馏水洗至溶液呈中性、再依次分别用乙醇和氯仿清洗粗产品10次,以除去未充分反应的原料和催化剂,最后,200℃条件抽真空3小时下,得到paf-41。
(6)将具有不锈钢内芯的玻璃毛细管的一端(2cm)浸泡到浓度为2mol.l-1的氢氧化钠溶液中,然后,用浓度为2mol.l-1的盐酸溶液中和多余的氢氧根离子,最后,纤维用去离子水清洗在空气中干燥,得到表面羟基化的不锈钢纤维;
(7)将5mmol4,4-二氨基二苯醚完全溶解于20ml无水n,n-二甲基甲酰胺中。在冰水浴条件下,边搅拌边向上述溶液中缓慢加入5mmol均苯四甲酸二酐,得到辅助胶,即聚酰亚胺的预聚液。
(8)将表面羟基化的不锈钢纤维的羟基化的一端插入步骤7得到的辅助胶中,缓慢取出,再插入到步骤5得到的paf-41的粉末中,90℃干燥2min;重复数次,直到达到所需的涂层厚度约80μm.
(9)将步骤8得到的纤维置于250℃条件下老化1小时,得到paf-41涂层纤维。
实施例4
一种paf-41涂层纤维的制备方法,包括以下步骤:
(1)在氮气保护下,将无水氯化铝(3.5mmol)加入100ml的圆底烧瓶中,随后注入40ml无水chcl3,并在60℃搅拌3小时。
(2)将1,3,5-三苯胺(1.5mmol))溶于20ml无水chcl3。
(3)将步骤2溶液注入到步骤1体系中,并在60℃下搅拌反应24小时。
(4)反应结束,且体系温度降到室温之后,过滤,分别用盐酸(1mol/l),甲醇,丙酮洗涤清洗产品3次。随后再依次用乙醇,四氢呋喃,氯仿对产物进行索式提取,提取时间均为2天。
(5)在80℃条件抽真空12小时下,得到paf-41。
(6)聚二甲基硅氧烷溶于甲苯中至粘度适中,得到辅助胶。
(7)将具有不锈钢内芯的石英毛细管的一端(3cm)浸泡到浓度为2mol/l的氢氧化钠溶液中,然后,用浓度为2mol/l的盐酸溶液中和多余的氢氧根离子,最后,纤维用去离子水清洗在空气中干燥,得到表面羟基化的石英毛细管;
(8)经步骤7得到的石英毛细管的表面羟基化的一端插入到步骤6得到的辅助胶中,缓慢取出,再插入步骤5所制得paf-41的粉末中,然后60℃干燥2分钟。重复3次,直到达到所需的涂层厚度约60μm.
(9)将步骤8得到的纤维置于240℃条件下老化1小时,得到paf-41涂层纤维。
实施例5
一种paf-41涂层纤维的制备方法,包括以下步骤:
(1)在氮气保护下,将无水fecl3(4.5mmol)加入100ml的圆底烧瓶中,随后注入40ml无水chcl3,并在60℃搅拌3小时。
(2)将1,3,5-三苯胺(1.5mmol))溶于20ml无水chcl3。
(3)将步骤2溶液注入到步骤1体系中,并在60℃下搅拌反应24小时。
(4)反应结束,且体系温度降到室温之后,过滤,分别用盐酸(1mol·l-1),甲醇,丙酮洗涤清洗产品3次。随后再依次用乙醇,四氢呋喃,氯仿对产物进行索式提取,提取时间均为2天。
(5)在80℃条件抽真空12小时下,得到paf-41。
(6)将具有不锈钢内芯的石英毛细管的一端(3cm)浸泡到浓度为2mol.l-1的氢氧化钠溶液中,然后,用浓度为2mol.l-1的盐酸溶液中和多余的氢氧根离子,最后,纤维用去离子水清洗在空气中干燥,得到表面羟基化的石英毛细管;
(7)将甲基三甲氧基硅烷300μl,羟基端头的聚二甲基硅氧烷180mg和聚甲基氢硅氧烷30mg加入离心管中,涡旋震荡5分钟。随后加入150μl三氟乙酸(95%),再次涡旋震荡5分钟。
(8)经步骤6得到的石英毛细管的表面羟基化的一端插入到步骤7得到的混合物中5分钟后,缓慢取出,再插入到步骤5得到的paf-41的粉末中,60℃干燥2分钟。重复3次,直到达到所需的涂层厚度约60μm.
(9)将步骤8得到的纤维置于240℃条件下老化1小时,得到paf-41涂层纤维。
实施例6
一种paf-41涂层纤维的制备方法,包括以下步骤:
(1)在氮气保护下,将无水cucl2(0.8mmol)加入200ml的圆底烧瓶中,随后注入40ml无水chcl3,并在75℃搅拌3小时。
(2)将1,3,5-三苯胺(10mmol))溶于50ml无水chcl3。
(3)将步骤2溶液注入到步骤1体系中,并在70℃下搅拌反应72小时。
(4)反应结束,且体系温度降到室温之后,过滤,分别用盐酸(3mol·l-1),甲醇,丙酮洗涤清洗产品3次。随后再依次用乙醇,四氢呋喃,氯仿对产物进行索式提取,提取时间均为1天。
(5)在80℃条件抽真空12小时下,得到paf-41。
(6)玻璃纤维的一端(1cm)浸泡到浓度为1.5mol.l-1的氢氧化钠溶液中,然后,用浓度为1.5mol.l-1的盐酸溶液中和多余的氢氧根离子,最后,纤维用去离子水清洗在空气中干燥,得到表面羟基化的玻璃纤维;
(7)将将4mmol4,4-二氨基二苯醚完全溶解于15ml无水n,n-二甲基甲酰胺中。在冰水浴条件下,边搅拌边向上述溶液中缓慢加入4mmol均苯四甲酸二酐,得到辅助胶,即聚酰亚胺的预聚液。
(8)经步骤6得到的玻璃纤维的表面羟基化的一端插入到步骤7得到的辅助胶中0.5分钟后,缓慢取出,再插入到步骤5得到的paf-41的粉末中,50℃干燥2分钟。重复2次,直到达到所需的涂层厚度约20μm.
(9)将步骤8得到的纤维置于200℃条件下老化5小时,得到paf-41涂层纤维。
实施例7
一种paf-41涂层纤维的制备方法,包括以下步骤:
(1)在氮气保护下,将无水氯化铝(6mmol)加入100ml的圆底烧瓶中,随后注入30ml无水chcl3,并在90℃搅拌3小时,然后转移到1000ml的圆底烧瓶中。
(2)将1,3,5-三苯胺(10mmol))溶于500ml无水chcl3。
(3)将步骤2溶液注入到步骤1体系中,并在60℃下搅拌反应24小时。
(4)反应结束,且体系温度降到室温之后,过滤,分别用盐酸(5mol·l-1),甲醇,丙酮洗涤清洗产品3次。随后再依次用乙醇,四氢呋喃,氯仿对产物进行索式提取,提取时间均为3天。
(5)在80℃条件抽真空12小时下,得到paf-41。
(6)将石英纤维一端(1cm)浸泡到浓度为0.5mol·l-1的氢氧化钠溶液中,然后,用浓度为0.5mol·l-1的盐酸溶液中和多余的氢氧根离子,最后,纤维用去离子水清洗在空气中干燥,得到表面羟基化的石英纤维;
(7)将聚二甲基硅氧烷溶解到甲苯中至粘度适中,得到辅助胶。
(8)经步骤6得到的石英纤维的表面羟基化的一端插入到步骤7得到的辅助胶中30分钟后,缓慢取出,再插入到步骤5得到的paf-41的粉末中,90℃干燥2分钟。重复数次,直到达到所需的涂层厚度约100μm.
(9)将步骤8得到的纤维置于230℃条件下老化3小时,得到paf-41涂层纤维。
实施例8
一种尿液中苯丙胺类物质的spme-gc检测方法,包括以下步骤:
(1)浓度为1.0mg/ml苯丙胺和甲基苯丙胺溶液用水稀释至10,50,100,500μg/l,作为标准工作溶液;
(2)取5ml标准工作溶液,浓度由低至高依次测试,,置于顶空瓶中,用naoh将溶液ph值调至14,加入1.5mgnacl,搅拌均匀,将顶空瓶封口;
(3)将实施例1所制备的paf-41涂层纤维连接到5μl的微量进样针上,作为固相微萃取纤维;
(4)将(3)中提到的固相微萃取纤维插入顶空瓶,推出固相微萃取纤维头,置于样品溶液顶空位置,在40℃条件下,保持30min,搅拌速度为600r/min;
(5)将萃取纤维头抽回微量注射器中,随后将微量注射器拔出,立刻插入气相色谱进样口,再次推出萃取头,进样口温度为240℃,解释时间为0.5min;
(6)经气相色谱分离,离子火焰检(fid)测器检测,将测得的峰面积(y)与标准溶液的质量浓度(x)进行线性回归分析,得到各目标化合物的标准曲线方程。所述的色谱条件为:db-5msui型毛细管柱(30m×0.25mmi.d.×0.25μm)。载气he流速为1.0ml/min,碰撞气n2流量为40ml/min.合成气(n2:o2=79:21)流速为40ml/min,氢气流速为400ml/min。检测器温度250℃,无分离模式2分钟。升温程序:70℃起始温度以10℃/min速度升至250℃保持10min。
(7)取含有苯丙胺的尿液样品1ml,用水稀释至5ml,按照上述1-6步骤中标准工作溶液的测试方法,对尿液样品进行测定,以保留时间定性,以测得目标物峰面积值,代入标准曲线方程,求得样品中的苯丙胺和甲基苯丙胺的含量。
具体结果见表1和表2,色谱图见图1。
表1方法的线性范围、相关系数、检出限和重复性。
表2实际样品测定和回收率
实施例9
一种尿液中苯丙胺类物质的spme-gc检测方法,包括以下步骤:
(1)浓度为1.0mg/ml苯丙胺和甲基苯丙胺溶液用水稀释至10,50,100,500μg/l,作为标准工作溶液;
(2)取8ml标准工作溶液,浓度由低至高依次测试,置于顶空瓶中,用naoh将溶液ph值调至14,加入2.4mgnacl,搅拌均匀,将顶空瓶封口;
(3)将实施例1所制备的paf-41涂层纤维连接到5μl的微量进样针上,作为固相微萃取纤维;
(4)将(3)中提到的固相微萃取纤维插入顶空瓶,推出固相微萃取纤维头,置于样品溶液顶空位置,在40℃条件下,保持20min,搅拌速度为400r/min;
(5)将萃取纤维头抽回微量注射器中,随后将微量注射器拔出,立刻插入气相色谱进样口,再次推出萃取头,进样口温度为220℃,解释时间为3min;
(6)经气相色谱分离,离子火焰检(fid)测器检测,将测得的峰面积(y)与标准溶液的质量浓度(x)进行线性回归分析,得到各目标化合物的标准曲线方程。所述的色谱条件为:db-5msui型毛细管柱(30m×0.25mmi.d.×0.25μm)。载气he流速为1.0ml/min,碰撞气n2流量为40ml/min.合成气(n2:o2=79:21)流速为40ml/min,氢气流速为400ml/min。检测器温度250℃,无分离模式2分钟。升温程序:70℃起始温度以10℃/min速度升至250℃保持10min。
(7)取含有苯丙胺的尿液样品1ml,用水稀释至8ml,按照上述1-6步骤中标准工作溶液的测试方法,对尿液样品进行测定,以保留时间定性,以测得目标物峰面积值,代入标准曲线方程,求得样品中的苯丙胺和甲基苯丙胺的含量。
具体结果见表3和表4。
表3方法的线性范围、相关系数、检出限和重复性。
表4实际样品测定和回收率
实施例10
一种尿液中苯丙胺类物质的spme-gc检测方法,包括以下步骤:
(1)浓度为1.0mg/ml苯丙胺和甲基苯丙胺溶液用水稀释至所需浓度作为标准溶液((10,50,100,500μg/l));
(2)取10ml标准工作溶液,浓度由低至高依次测试,置于顶空瓶中,用naoh将溶液ph值调至14,加入3mgnacl,搅拌均匀,将顶空瓶封口;
(3)将实施例4所制备的paf-41涂层纤维连接到5μl的微量进样针上,作为固相微萃取装置纤维;
(4)将(3)中提到的固相微萃取纤维插入顶空瓶,推出固相微萃取纤维头,置于样品溶液顶空位置,在40℃条件下,保持60min,搅拌速度为500r/min;
(5)将萃取纤维头抽回微量注射器中,随后将微量注射器拔出,立刻插入气相色谱进样口,再次推出萃取头,进样口温度为250℃,解释时间为1min;
(6)经气相色谱分离,离子火焰检(fid)测器检测,将测得的峰面积(y)与标准溶液的质量浓度(x)进行线性回归分析,得到各目标化合物的标准曲线方程。,所述的色谱条件为:db-5msui型毛细管柱(30m×0.25mmi.d.×0.25μm)。载气he流速为1.0ml/min,碰撞气n2流量为40ml/min.合成气(n2:o2=79:21)流速为40ml/min,氢气流速为400ml/min。检测器温度250℃,无分离模式2分钟。升温程序:70℃起始温度以10℃/min速度升至250℃保持10min。
(7)取含有苯丙胺的尿液样品1ml,用水稀释至10ml,按照上述1-6步骤中标准工作溶液的测试方法,对尿液样品进行测定,以保留时间定性,以测得目标物峰面积值,代入标准曲线方程,求得样品中的苯丙胺和甲基苯丙胺的含量。
具体结果见表5和表6。
表5方法的线性范围、相关系数、检出限和重复性。
表6实际样品测定和回收率
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!