用于筛查MPSI、II、IIIA、IIIB、IVA、VI和VII的试剂和方法与流程
本申请是申请日为2014年9月5日、申请号为201480056093.8、发明名称为“用于筛查mpsi、ii、iiia、iiib、iva、vi和vii的试剂和方法”的中国发明专利申请的分案申请。
相关申请的交叉引用
本申请要求2013年9月5日提交的美国临时申请号61/874,293、2013年9月5日提交的美国临时申请号61/874,331、2013年9月9日提交的美国临时申请号61/960,102、2013年9月9日提交的美国临时申请号61/960,113、2014年3月7日提交的美国临时申请号61/949,970、2014年3月20日提交的美国临时申请号61/968,021和2014年6月13日提交的美国临时申请号62/012,020的权益,每篇明确地通过引用整体并入本文。
政府许可权利的声明
本发明在国立卫生研究院授予的授权编号dk67859下在政府支持下完成。美国政府具有本发明的某些权利。
本发明涉及用于筛查mpsi、ii、iiia、iiib、iva、vi和vii的试剂、方法和试剂盒。
背景技术:
用于溶酶体贮积障碍(lsd)亚类的治疗已经变得可行,且在许多情况下,治疗的及早开始会导致临床改善。这些鼓舞人心的结果已经在lsd的新生儿筛查中产生广泛分布的兴趣。
已经确立新生儿筛查程序来量化与这些可治疗的疾病有关的代谢物的水平。纽约州(newyorkstate)现在提供克拉伯病筛查,并且关于lsd扩张的新生儿筛查的新近立法已经在几个其它州中通过,并在台湾进行关于庞皮病和法布里病的新生儿筛查。
mps(mpsi至vii)是由降解糖胺聚糖(包括类肝素(heparan)、皮肤素、角质素或硫酸软骨素)的溶酶体酶之一的缺乏造成的一组代谢疾病/综合征。有关的酶包括五种硫酸酯酶、四种外切糖苷酶和一种非水解的乙酰基-n-转移酶。这些综合征导致在溶酶体中积聚未降解的或部分地降解的糖胺聚糖,从而导致不可逆的多系统器官损伤。
尽管治疗最近已经变得可用于一些mps综合征,来自这些治疗的最佳益处要求在不可逆的症状发作之前开始治疗。mps综合征的及早检测会使治疗的潜在益处最大化,因而需要开发适用于及早诊断的试验。同样需要开发快速的、廉价的和可靠的诊断程序,其使用干燥血斑点(dbs)作为样品来源,诸如呈送给新生儿筛查实验室的那些。
因此,需要用于溶酶体酶活性的新生儿筛查的方法和试剂,特别是允许改善mpsi、ii、iiia、iiib、iva、vi和vii的筛查的方法和试剂。本发明满足了该需要并提供了其它有关的优点。
技术实现要素:
本发明提供了用于筛查mpsi、ii、iiia、iiib、iva、vi和vii的试剂,用于筛查mpsi、ii、iiia、iiib、iva、vi和vii的方法,和包括所述试剂的试剂盒。
在一个方面,本发明提供了用于测定与溶酶体贮积病有关的一种或多种酶的方法。
在第一个实施方案中,所述方法包括:
(a)使样品与第一溶液接触以提供包含一种或多种溶酶体酶的溶液;
(b)使溶液中的一种或多种溶酶体酶与要分析的每种溶酶体酶的酶底物接触,和将所述底物与所述酶一起温育足以提供包含所述样品中存在的每种溶酶体酶的酶产物的溶液的时间,
其中每种溶酶体酶的酶底物是具有碳水化合物部分和糖苷配基部分且具有下式的化合物:
其中s是这样的碳水化合物部分,其当与所述糖苷配基部分共价地偶联时提供选自以下的酶的底物:
(i)α-l-艾杜糖醛酸酶(alpha-l-iduronidase);
(ii)艾杜糖醛酸2-硫酸酯酶(iduronate2-sulfatase);
(iii)类肝素n-硫酸酯酶;
(iv)n-乙酰基-α-d-氨基葡糖苷酶;
(v)n-乙酰基半乳糖胺6-硫酸酯(sulfate)-硫酸酯酶;
(vi)n-乙酰基半乳糖胺4-硫酸酯-硫酸酯酶;和
(vii)β-葡糖醛酸糖苷酶;
l2是包含1-20个碳原子的连接体(linker),其中一个或多个碳原子可以被选自n、o和s的杂原子替换,和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
l3是包含1-20个碳原子的连接体,其中一个或多个碳原子可以被选自n、o或s的杂原子替换,和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
l4是任选的,且当存在时是包含1-20个碳原子的连接体,其中一个或多个碳原子可以被选自n、o或s的杂原子替换),和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
r1是c1-c10烷基或c1-c10烷氧基;
r2在每次出现时独立地选自c1-c10烷基、c1-c10烷氧基、卤素、硝基、-c(=o)nhr或-c(=o)or,其中r是c1-c8烷基;
r3是c1-c10烷基或被取代的或未被取代的c6-c10芳基;且
n是0、1、2、3或4;和
(c)确定所述酶产物中的一种或多种的量。
在某些实施方案中,所述方法进一步包括使所述酶产物与糖原水解酶(glycohydrolase)接触以提供第二酶产物。在某些实施方案中,所述方法进一步包括添加抑制剂以阻断作用于n-乙酰基半乳糖胺6-硫酸酯-硫酸酯酶或n-乙酰基半乳糖胺4-硫酸酯-硫酸酯酶的底物的内源性糖原水解酶活性。
在第二个实施方案中,所述方法包括:
(a)使样品与第一溶液接触以提供包含一种或多种溶酶体酶的溶液;
(b)使溶液中的一种或多种溶酶体酶与要分析的每种溶酶体酶的酶底物接触,和将所述底物与所述酶一起温育足以提供包含存在于样品中的每种溶酶体酶的第一酶产物的溶液的时间;
(c)使所述第一酶产物遇到糖原水解酶以提供易于通过糖原水解酶实现的进一步酶促反应的每种第一酶产物的第二酶产物;和
(d)确定一种或多种第一酶产物和/或一种或多种第二酶产物的量。
在以上方法中,某些第一酶产物不易于通过糖原水解酶实现进一步酶促反应。不易于通过糖原水解酶实现的进一步酶促反应的第一酶产物由选自以下的酶的作用提供:
(a)α-l-艾杜糖醛酸酶;
(b)n-乙酰基-α-d-氨基葡糖苷酶;和
(c)β-葡糖醛酸糖苷酶。
易于通过糖原水解酶实现的进一步酶促反应的第一酶产物由选自以下的酶的作用提供:
(a)艾杜糖醛酸2-硫酸酯酶;
(b)类肝素n-硫酸酯酶;
(c)n-乙酰基半乳糖胺6-硫酸酯-硫酸酯酶;和
(d)n-乙酰基半乳糖胺4-硫酸酯-硫酸酯酶。
在某些实施方案中,所述方法进一步包括添加抑制剂以阻断作用于n-乙酰基半乳糖胺6-硫酸酯-硫酸酯酶或n-乙酰基半乳糖胺4-硫酸酯-硫酸酯酶的底物的内源性糖原水解酶活性,其中所述抑制剂不会显著地抑制步骤(c)的糖原水解酶的活性。
在以上方法中,所述一种或多种溶酶体酶包含选自以下的酶:
(a)α-l-艾杜糖醛酸酶;
(b)艾杜糖醛酸2-硫酸酯酶;
(c)类肝素n-硫酸酯酶;
(d)n-乙酰基-α-d-氨基葡糖苷酶;
(e)n-乙酰基半乳糖胺6-硫酸酯-硫酸酯酶;
(f)n-乙酰基半乳糖胺4-硫酸酯-硫酸酯酶;和
(g)β-葡糖醛酸糖苷酶。
在以上方法的某些实施方案中,在使溶酶体酶与底物接触之前、之后或同时加入要分析的每种溶酶体酶的内部标准品。
在以上方法的某些实施方案中,在确定所述酶产物中的一种或多种的量之前猝灭酶反应。
在以上方法的实施方案中,所述样品是血液或组织样品。在某些实施方案中,所述样品是干燥血斑点。
代表性的糖原水解酶包括人己糖胺酶a、细菌n-乙酰基己糖胺酶、细菌β-n-乙酰基半乳糖胺酶、α-l-艾杜糖醛酸酶、β-半乳糖苷酶(曲霉菌属)和α-葡萄糖苷酶(酵母)。
代表性的抑制剂包括(z)-o-(2-乙酰氨基-2-脱氧-d-吡喃葡萄糖亚基(glucopyranosylidene))-氨基n-苯基氨基甲酸酯、1-脱氧野尻霉素(nojirmycin)、栗精胺、苦马豆素、打碗花精b2、isofagamine、达菲、葡糖酸肟基内酯(gluconohydroximolactone)、葡糖醛酸和它的内酯和内酰胺、扎那米韦、米格列醇、苯乙基取代的葡萄糖-和半乳糖-咪唑、n-羟基乙基脱氢野尻霉素、galnac噻唑啉和glcnac噻唑啉。
在以上方法的某些实施方案中,确定酶产物(例如,第一和/或第二酶产物)的量包括质谱分析。在某些实施方案中,确定酶产物的量包括通过质谱分析来确定每种产物与它的内部标准品的比率。在某些实施方案中,确定酶产物的量包括串联质谱分析,其中产生产物和它们的内部标准品的母离子,分离,并进行碰撞诱导的解离以提供产物片段离子和内部标准品片段离子。在某些实施方案中,确定酶产物的量包括对比产物片段离子和内部标准品片段离子的峰强度以计算产物的量。在某些实施方案中,确定酶产物的量包括通过液相色谱法或通过流动注射将产物引导至质谱仪。
在以上方法的某些实施方案中,确定酶产物的量包括荧光分析。
在以上方法的某些实施方案中,所述方法进一步包括使用酶产物的量来确定所述样品是否来自用于治疗与一种或多种溶酶体酶缺乏有关的病症的候选物。
在上面指出的方法的第二个实施方案中,在某些实施方案中,所述底物具有碳水化合物部分和糖苷配基部分且具有下式:
其中s是这样的碳水化合物部分,其当与所述糖苷配基部分共价地偶联时提供选自以下的酶的底物:
(a)α-l-艾杜糖醛酸酶;
(b)艾杜糖醛酸2-硫酸酯酶;
(c)类肝素n-硫酸酯酶;
(d)n-乙酰基-α-d-氨基葡糖苷酶;
(e)n-乙酰基半乳糖胺6-硫酸酯-硫酸酯酶;
(f)n-乙酰基半乳糖胺4-硫酸酯-硫酸酯酶;和
(g)β-葡糖醛酸糖苷酶;
l2是包含1-20个碳原子的连接体,其中一个或多个碳原子可以被选自n、o和s的杂原子替换,和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
l3是包含1-20个碳原子的连接体,其中一个或多个碳原子可以被选自n、o或s的杂原子替换,和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
l4是任选的,且当存在时是包含1-20个碳原子的连接体,其中一个或多个碳原子可以被选自n、o或s的杂原子替换),和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
r1是c1-c10烷基或c1-c10烷氧基;
r2在每次出现时独立地选自c1-c10烷基、c1-c10烷氧基、卤素、硝基、-c(=o)nhr或-c(=o)or,其中r是c1-c8烷基;
r3是c1-c10烷基或被取代的或未被取代的c6-c10芳基;且
n是0、1、2、3或4。
在本发明的方法中有用的代表性底物包括本文描述的和下面指出的本发明的底物。
在本发明的方法中有用的代表性内部标准品包括本文描述的本发明的内部标准品。
在本发明的另一个方面,提供了用于测定与溶酶体贮积病有关的一种或多种酶的试剂(底物和内部标准品)。
代表性的底物包括具有碳水化合物部分和糖苷配基部分且具有下式的化合物:
其中
s是这样的碳水化合物部分,其当与所述糖苷配基部分共价地偶联时提供选自以下的酶的底物:
(a)α-l-艾杜糖醛酸酶;
(b)艾杜糖醛酸2-硫酸酯酶;
(c)类肝素n-硫酸酯酶;
(d)n-乙酰基-α-d-氨基葡糖苷酶;
(e)n-乙酰基半乳糖胺6-硫酸酯-硫酸酯酶;
(f)n-乙酰基半乳糖胺4-硫酸酯-硫酸酯酶;和
(g)β-葡糖醛酸糖苷酶;
l2是包含1-20个碳原子的连接体,其中一个或多个碳原子可以被选自n、o和s的杂原子替换,和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
l3是包含1-20个碳原子的连接体,其中一个或多个碳原子可以被选自n、o或s的杂原子替换,和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
l4是任选的,且当存在时是包含1-20个碳原子的连接体,其中一个或多个碳原子可以被选自n、o或s的杂原子替换),和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
r1是c1-c10烷基或c1-c10烷氧基;
r2在每次出现时独立地选自c1-c10烷基、c1-c10烷氧基、卤素、硝基、-c(=o)nhr或-c(=o)or,其中r是c1-c8烷基;
r3是c1-c10烷基或被取代的或未被取代的c6-c10芳基;且
n是0、1、2、3或4。
在某些实施方案中,l2是-(ch2)n-,其中n是1-6。
在某些实施方案中,l3是-(ch2)m-,其中m是1-12。
在某些实施方案中,l4是-(ch2)n-,其中n是1-6。
在某些实施方案中,l4不存在。
在某些实施方案中,r1是c1-c5烷基。
在某些实施方案中,r2是c1-c8烷基。
在某些实施方案中,r3是c1-c6烷基。
在某些实施方案中,r3是苯基。
代表性的底物显示在下面。
在某些实施方案中,所述底物具有下式:
在一个实施方案中,所述底物具有下式:
在某些实施方案中,所述底物具有下式:
在一个实施方案中,所述底物具有下式:
在某些实施方案中,所述底物具有下式:
在一个实施方案中,所述底物具有下式:
在某些实施方案中,所述底物具有下式:
在一个实施方案中,所述底物具有下式:
在某些实施方案中,所述底物具有下式:
在一个实施方案中,所述底物具有下式:
在某些实施方案中,所述底物具有下式:
在一个实施方案中,所述底物具有下式:
在某些实施方案中,所述底物具有下式:
在一个实施方案中,所述底物具有下式:
在本发明的另一个方面,提供了用于测定与溶酶体贮积病有关的一种或多种酶的试剂盒。在一个实施方案中,所述试剂盒包括本发明的一种或多种试剂(例如,底物和内部标准品)。在某些实施方案中,能够用所述试剂盒测定的酶包括α-l-艾杜糖醛酸酶(mps-i)、艾杜糖醛酸-2-硫酸酯酶(mps-ii)、类肝素n-硫酸酯酶(mps-iiia)、n-乙酰基-α-d-氨基葡萄糖苷酶(glycosaminidase)(mps-iiib)、n-乙酰基半乳糖胺-6-硫酸酯-硫酸酯酶(mps-iva)、n-乙酰基半乳糖胺-4-硫酸酯-硫酸酯酶(mps-vi)和β-葡糖醛酸糖苷酶(mps-vii)中的一种或多种。
附图说明
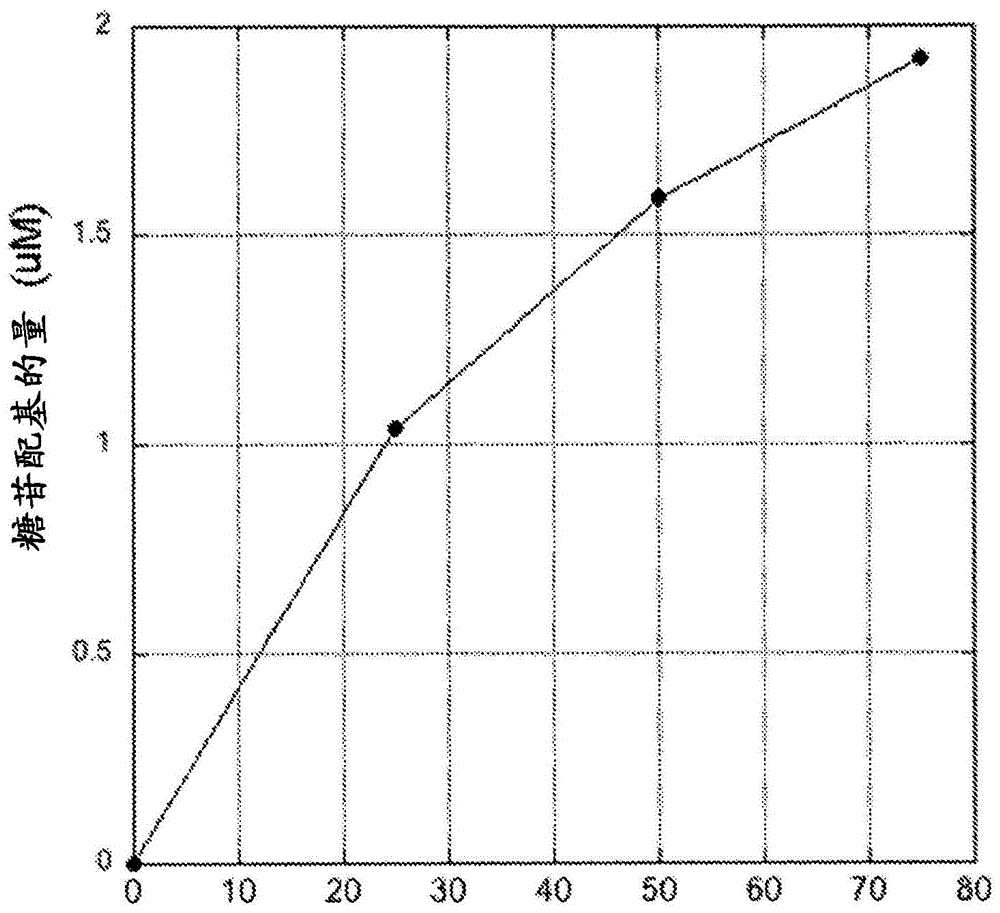
图1的图解释了随着在本发明的代表性mps-vi测定中加入的己糖胺酶a的量而变化的释放的糖苷配基的量。通过uhplc-ms/ms来检测糖苷配基。
图2是本发明的代表性mps-iva底物的制备的示意图。
图3是本发明的代表性mps-vi底物的制备的示意图。
图4是本发明的代表性mps-vi底物的制备的示意图。
图5是代表性mps-vi产物的制备的示意图。
发明详述
本发明提供了用于筛查粘多糖累积病i、ii、iiia、iiib、iva、vi和vii的试剂(分别是mps-i、ii、iiia、iib、iva、vi和vii),用于筛查mps-i、ii、iiia、iiib、iva、vi和vii的方法,和包括所述试剂的试剂盒。
在一个方面,本发明提供了用于筛查mps-i、ii、iiia、iiib、iva、vi和vii筛查的试剂。在某些实施方案中,本发明的试剂包括用于筛查mpsi、ii、iiia、iiib、iva、vi和vii的底物(s)、产物(p)和内部标准品(is)。
在另一个方面,本发明提供了用于筛查mpsi、ii、iiia、iiib、iva、vi和vii的方法。所述方法测定特定酶,所述酶的缺乏会导致溶酶体贮积病状况。所述方法有利地测定α-l-艾杜糖醛酸酶(mps-i)、艾杜糖醛酸-2-硫酸酯酶(mps-ii)、类肝素n-硫酸酯酶(mps-iiia)、n-乙酰基-α-d-氨基葡萄糖苷酶(mps-iiib)、n-乙酰基半乳糖胺-6-硫酸酯-硫酸酯酶(mps-iva)、n-乙酰基半乳糖胺-4-硫酸酯-硫酸酯酶(mps-vi)和β-葡糖醛酸糖苷酶(mps-vii)中的一种或多种。
试剂
在一个方面,本发明提供了可以有利地用于测定酶的试剂。所述试剂包括酶底物(s)、酶产物(p)和测定内部标准品(is)。在某些实施方案中,将一种或多种底物(s)和它们的对应内部标准品(is)与合适的酶来源(诸如来自新生儿筛查卡或尿样品的干燥血斑点)一起在合适的缓冲液中温育足够的时间以形成一种或多种产物(p),随后通过串联质谱法检测所述产物。在某些实施方案中,除了所述标准品具有不同的质量(例如,同系物或重同位素取代的,诸如氘和/或碳-13取代)以外,所述内部标准品(is)在化学上类似于或等同于酶形成的产物。在其它实施方案中,将一种或多种底物(s)与合适的酶来源一起在合适的缓冲液中温育以形成一种或多种产物(p),随后通过荧光分析来检测所述产物。
有利地用本发明的试剂测定的酶包括以下酶:
(a)α-l-艾杜糖醛酸酶,其作用于底物mps-i-s以产生产物mps-i-p,且所述测定利用内部标准品mps-i-is;
(b)艾杜糖醛酸-2-硫酸酯酶,其作用于底物mps-ii-s以产生产物mps-ii-p,且所述测定利用内部标准品mps-ii-is;
(c)类肝素n-硫酸酯酶,其作用于底物mps-iiia-s以产生产物mps-iiia-p,且所述测定利用内部标准品mps-iiia-is;
(d)n-乙酰基-α-d-氨基葡萄糖苷酶,其作用于底物mps-iiib-s以产生产物mps-iiib-p,且所述测定利用内部标准品mps-iiib-is;
(e)n-乙酰基半乳糖胺-6-硫酸酯-硫酸酯酶,其作用于底物mps-iva-s以产生产物mps-iva-p,且所述测定利用内部标准品mps-iva-is;
(f)n-乙酰基半乳糖胺-4-硫酸酯-硫酸酯酶,其作用于底物mps-vi-s以产生产物mps-vi-p,且所述测定利用内部标准品mps-vi-is;和
(g)β-葡糖醛酸糖苷酶,其作用于底物mps-vii-s以产生产物mps-vii-p,且所述测定利用内部标准品mps-vii-is。
以下是针对mps-i、mps-ii、mps-iiia、mps-iiib、mpsiva、mps-vi和mps-vii的本发明的试剂、底物(s)、产物(p)和内部标准品(is)的描述。
糖-糖苷配基.本发明的底物是糖苷。术语“糖苷”表示这样的化合物,其中糖基(糖苷配糖基)通过它的异头碳用糖苷键键合至另一个基团(糖苷配基)。
本发明的底物被表征为具有糖-糖苷配基结构。所述底物的糖组分要么是作为特定酶的底物的天然糖,要么是维持足以成为要测定的特定酶的底物的功能的改性糖。所述底物的糖苷配基组分允许分析酶活性。所述底物的糖苷配基组分也是要被分析以确定酶活性的酶产物的组分。所述糖苷配基组分包括用于质谱法或荧光分析的官能团(functionality)。当通过质谱法分析时,可以采用具有不同于产物的质量的内部标准品。所述内部标准品要么在结构上与产物相同且包括一个或多个同位素(例如,氘或13c),要么在结构上类似且具有在功能上等效的结构和结构变异(例如,同系物:-(ch2)5-相对于-(ch2)6-,或反之亦然)。
糖苷配基.本发明的试剂包括糖苷配基组分。糖苷配基的性质可以随用于测定目标酶的分析技术的性质而变化。代表性糖苷配基由下面的式(i)-(vi)表示。在下面的糖苷配基结构中,波浪线描绘了与糖异头碳的连接点。
在某些实施方案中,所述糖苷配基是具有下式的a型糖苷配基:
其中l1是将g1共价地偶联至香豆素部分的连接体,且其中x1、x2、x3和x4在每次出现时独立地是氢或卤素(例如,氯)。
在某些实施方案中,l1包括1-20个碳原子(支链或直链),其中一个或多个碳原子可以被醚氧或-c(=o)o-基团、硫醚硫或-c(=o)s-基团、nh、n(r)或-c(=o)nh-或-c(=o)nr-(其中r是1-6个碳的烷基)替换。一个或多个碳原子氢原子的取代是任选的。在某些实施方案中,l1是-ch2-c(=o)-nh-(ch2)5-g1。
g1包括带正电荷的基团(例如,永久带正电荷的基团诸如季铵离子)诸如下述之一:
(a)n(ra)(rb)(rc)+,其中ra、rb和rc各自独立地是h或1-6个碳的烷基;
(b)s(ra)(rb)+,其中ra和rb如上;
(c)以下类型的吡啶
(d)以下类型的吡啶
(e)以下类型的吡啶
(f)以下类型的吡啶
在某些实施方案中,l1是-ch2-c(=o)-nh-(ch2)5-c(=o)nh-ch2-c6h4-n+(c5h5),其中-c6h4-n+(c5h5)是对-吡啶
应当理解,除了上面定义的香豆素(伞形酮)糖苷配基以外,可以利用其它荧光糖苷配基(例如,荧光素、试卤灵、罗丹明、硝基苯酚和7-羟基-9h-(1,3-二氯-9,9-二甲基吖啶-2-酮和它们的卤代衍生物),如下所述。
在某些实施方案中,所述a型糖苷配基具有下式:
其中rd是氢或甲基,且x1、x2、x3和x4在每次出现时独立地是氢或卤素(例如,氯)。除了上面定义的香豆素(伞形酮)糖苷配基以外,应当理解,可以利用其它荧光糖苷配基。合适的其它糖苷配基包括荧光素、试卤灵、罗丹明、硝基苯酚和7-羟基-9h-(1,3-二氯-9,9-二甲基吖啶-2-酮和它们的卤代衍生物。对于荧光素、试卤灵、硝基苯酚和7-羟基-9h-(1,3-二氯-9,9-二甲基吖啶-2-酮,糖苷配基通过它的羟基偶联至糖,如在上面指出的香豆素中。对于罗丹明,糖苷配基通过它的氨基偶联至糖。
a型糖苷配基组分可以被包括在本发明的试剂中以赋予荧光官能团(functionality)(即,香豆素、伞形酮、荧光素、试卤灵、硝基苯酚、罗丹明、7-羟基-9h-(1,3-二氯-9,9-二甲基吖啶-2-酮部分)和提供可以通过荧光技术来分析的试剂。
在一个实施方案中,所述糖苷配基是b型糖苷配基且具有下式:
l2包括1-20个碳原子(支链或直链),其中一个或多个碳原子可以被杂原子(例如,n、o、s)替换和/或一个或多个碳原子可以被取代(例如,c1-c6烷基、卤素)。在某些实施方案中,l2是-(ch2)n-,其中n是1-6。在某些实施方案中,l2是-(ch2)2-。
l3包括1-20个碳原子(支链或直链),其中一个或多个碳原子可以被杂原子(例如,n、o、s)替换和/或一个或多个碳原子可以被取代(例如,c1-c6烷基、卤素)。在某些实施方案中,l3是-(ch2)m-,其中m是1-12。在某些实施方案中,l3是-(ch2)m-,其中m是4、5或6。
r1是c1-c10烷基(例如,支链或直链)或c1-c10烷氧基(例如,otbu)。在某些实施方案中,r1是c1-c5烷基(例如,甲基、乙基、正丙基、正丁基、正戊基)。
r2在每次出现时独立地选自c1-c10烷基(例如,支链或直链)、c1-c10烷氧基(例如,支链或直链)、卤素(例如,氟、氯)、硝基、-c(=o)nhr或-c(=o)or,其中r是c1-c8烷基(例如,甲基),且n是0、1、2、3或4。r2的代表性取代型式(相对于酚氧)包括2-、2,6-二、3-、3,5-二、和2,3-二(即,2-和6-位是邻位,3-和5-位是间位)。在某些实施方案中,r2是位于酚氧的邻位或间位的氟、甲基或甲氧基(例如,2-氟、2-甲基、2-甲氧基、3-氟、3-甲基、3-甲氧基)。在其它实施方案中,r2是位于酚氧的间位的氟、甲基或甲氧基。在某些实施方案中,n是0,且所述亚苯基是未被取代的。
r3是c1-c10烷基(例如,支链或直链)或被取代的或未被取代的c6-c10芳基(例如,苯基)。芳基取代基包括c1-c10烷基(例如,支链或直链)和卤素(例如,氯)。在某些实施方案中,r3是c1-c6烷基(例如,乙基、正丙基、正丁基、正戊基)。在其它实施方案中,r3是苯基。
在某些实施方案中,所述b型糖苷配基具有下式:
其中l2、l3、r1、r2和n如上面关于式(iii)所述,且r4在每次出现时独立地选自c1-c6烷基(例如,甲基),且m是0、1、2、3、4或5。在某些实施方案中,m是0。在其它实施方案中,r4是c1-c5烷基(例如,甲基)且m是2。
在另一个实施方案中,所述b型糖苷配基具有下式:
其中l2、l3、r1、r2、r3和n如上面关于式(iii)所述,且l4包括1-20个碳原子(支链或直链),其中一个或多个碳原子可以被杂原子(例如,n、o、s)替换和/或一个或多个碳原子可以被取代(例如,c1-c6烷基、卤素)。在某些实施方案中,l4是-(ch2)n-,其中n是1-6。在某些实施方案中,l4是-(ch2)-。
在某些实施方案中,所述b型糖苷配基具有下式:
其中l2、l3、l4、r1、r2、r4、n和m如上面关于式(v)所述。
重原子衍生物.在某些实施方案中,本发明的试剂包括它们的重原子衍生物(即,包括一个或多个重原子同位素的衍生物)。所述重原子衍生物可用作利用质谱分析的测定的内部标准品。在某些实施方案中,a型和b型糖苷配基具有一个或多个(例如,三个或更多个)被氘替换的氢原子或者一个或多个(例如,三个或更多个)被碳-13替换的碳原子,使得糖苷配基的质量增加一个或多个道尔顿。如同在酶产物和内部标准品对(例如,mps-ii-p和mps-ii-is)之间一样,所述试剂在质量上不同且质量的差异可以通过使用额外的(或更少的)原子(例如,使化合物的一部分的长度改变一个或多个亚甲基,例如,对于l1、l2、l3、l4、r1、r2、r3或r4)或通过掺入重原子(例如,氘替代氢、13c替代碳、15n替代氮,例如,在l1、l2、l3、l4、r1、r2、r3或r4中)来实现。
对于mps-i、ii、iiia、iiib、iva、vi和vii试剂而言,底物/内部标准品对的代表性糖苷配基包括以下的:
对于mps-i底物(参考式(iv)),r1是甲基,r2是氢且n是4,l2是-ch2ch2-,l3是-(ch2)6-,且r4是氢且m是5;对于mps-i内部标准品,r1是甲基,r2是氢且n是4,l2是-ch2ch2-,l3是-(ch2)6-,且r4是氘且m是5;
对于mps-ii底物(参考式(iv)),r1是正丁基,r2是氢且n是4,l2是-ch2ch2-,l3是-(ch2)6-,且r4是氢且m是5;对于mps-ii内部标准品,r1是正丁基,r2是氢且n是4,l2是-ch2ch2-,l3是-(ch2)6-,且r4是氘且m是5;
对于mps-iiia底物(参考式(iv)),r1是乙基,r2是氢且n是4,l2是-ch2ch2-,l3是-(ch2)6-,且r4是氢且m是5;对于mps-iiia内部标准品,r1是乙基,r2是氢且n是4,l2是-ch2ch2-,l3是-(ch2)6-,r4是氘且m是5;
对于mps-iiib底物(参考式(iii)),r1是正丁基,r2是氢且n是4,l2是-ch2ch2-,l3是-(ch2)6-,且r3是乙基;对于mps-iiib内部标准品,r1是正丁基,r2是氘且n是4,l2是-ch2ch2-,l3是-(ch2)6-,r3是乙基;
对于mps-iva底物(参考式(iii)),r1是正丁基,r2是氢且n是4,l2是-ch2ch2-,l3是-(ch2)5-,且r3是3,5-二甲基苯基;对于mps-iva内部标准品,r1是正丁基,l2是-ch2ch2-,l3是-(ch2)5-,r3是3,5-二甲基苯基,且r2是氘且n是4;且在一个替代实施方案中,对于mps-iva底物,r1是正戊基,r2是氢且n是4,l2是-ch2ch2-,l3是-(ch2)6-,且r3是苯基;对于mps-iva内部标准品,r1是正戊基,r2是氢且n是4,l2是-ch2ch2-,l3是-(ch2)6-,r3是d5-苯基;
对于mps-vi底物(参考式(iv)),r1是正丁基,r2是氢且n是4,l2是-ch2ch2-,l3是-(ch2)5-,且r4是氢且m是5;对于mps-vi内部标准品,r1是正丁基,r2是氢且n是4,l2是-ch2ch2-,l3是-(ch2)5-,且r4是氘且m是5;和
对于mps-vii底物(参考式(iii)),r1是丁基,r2是氢且n是4,l2是-ch2ch2-,l3是-(ch2)6-,且r3是丙基;对于mps-vii内部标准品,r1是丁基,r2是氘且n是4,l2是-ch2ch2-,l3是-(ch2)6-,且r3是丙基。
盐.在某些实施方案中,所述试剂包括氨基(例如,-nh2)、羧酸基(-co2h)、磺酸基(例如,-oso3h)和酰氨基磺酸基(例如,-nhso3h),其取决于ph环境可以变成带电荷的基团(例如,-nh3+、-co2-、-oso3-、-nhso3-)。应当理解,本发明的试剂包括它们的盐(例如,金属盐)。
在实施例1、4和5中描述了代表性mps-i、ii、iiia、iiib、iva和vi试剂的制备。
以下是本发明的代表性试剂(即,化合物)的描述。
mps-i试剂
在一个实施方案中,本发明提供了由下式定义的mps-i试剂(s、p和is试剂)、它的盐及其重原子衍生物:
其中
r1=糖苷配基,r2=h
或者
r1=h,r2=糖苷配基
r3=h、oh、nh2且r4=h
或者
r4=h、oh、nh2且r3=h
r5=h、oh、nh2且r6=h
或者
r6=h、oh、nh2且r5=h
r7=h、oh、nh2且r8=h
或者
r8=h、oh、nh2且r7=h
r9=cooh且r10=h
或者
r10=cooh且r9=h
前提条件是,r3和r4、r5和r6、以及r7和r8的对中仅一对可以具有每个r基团为氢(即,所述碳水化合物环可以在环中包括仅单个亚甲基(-ch2-))。
对于上面的化合物,糖苷配基如上所述。
在上面定义的mps-i试剂的某些实施方案中,所述碳水化合物部分被氢原子替换;在该情况下,将氢原子添加至糖苷配基。这些试剂是mps-i酶产物和内部标准品的代表。
在一个实施方案中,所述糖具有下式:
在某些实施方案中,所述化合物包括氨基(例如,-nh2)和羧酸基(-co2h),其随ph环境可以变成带电荷的基团(例如,-nh3+或-co2-)。应当理解,本发明的化合物包括它们的盐(例如,金属盐)。
如上面所指出的,本发明的化合物包括它们的重原子衍生物。所述重原子衍生物可用作内部标准品。在某些实施方案中,a型和b型糖苷配基具有一个或多个(例如,三个或更多个)被氘替换的氢原子或者一个或更多个(例如,三个或更多个)被碳-13替换的碳原子,使得糖苷配基的质量增加一个或多个道尔顿。所述酶产物和内部标准品在质量上不同且质量的差异可以通过使用额外的原子(例如,使化合物的一部分的长度改变一个或多个亚甲基)或通过掺入重原子(例如,氘替代氢、13c替代碳、15n替代氮)来实现。
在上面定义的mps-i试剂的某些实施方案中,所述碳水化合物部分被氢原子替换;在该情况下,将氢原子添加至糖苷配基。这些试剂是mps-i酶产物和内部标准品的代表。
代表性mps-i试剂包括下述化合物。
在某些实施方案中,mps-i底物具有下式:
其中l2、l3、r1、r2和n如上面关于式(iii)所述。
在某些实施方案中,mps-i底物具有下式:
其中l2、l3和r1如上面关于式(iii)所述。
代表性mps-i底物具有下式:
从以上底物(mps-i-s)形成的mps-i产物具有下式:
其中l2、l3、r1、r2和n如上面关于式(iii)所述。
代表性mps-i产物具有下式:
可用于测定从以上底物(mps-i-s)形成的产物的mps-i内部标准品具有下式:
其中l2、l3、r1、r2和n如上面关于式(iii)所述,且其中mps-i-is的质量不同于mps-i-p的质量,使得二者可通过质谱法辨别。如上面所指出的,mps-i-is可以包括一个或多个重原子同位素(在上面的结构中未显示),或可以具有结构变异(例如,底物的l2、l3、r1和r2中的一个或多个不同于内部标准品的l2、l3、r1和r2)。
代表性mps-i内部标准品具有下式:
使用代表性mps-i内部标准品可以测定从代表性mps-i底物衍生出的代表性mps-i产物。
mps-ii试剂
在一个实施方案中,本发明提供了由下式定义的mps-ii试剂(s、p和is试剂)、它的盐及其重原子衍生物:
其中
r1=糖苷配基,r2=h
或者
r1=h,r2=糖苷配基
r3=oso3h、nhso3h且r4=h
或者
r4=oso3h、nhso3h且r3=h
r5=h、oh、nh2且r6=h
或者
r6=h、oh、nh2且r5=h
r7=h、oh、nh2且r8=h
或者
r8=h、oh、nh2且r7=h
r9=cooh且r10=h
或者
r10=cooh且r9=h
前提条件是,r5和r6、以及r7和r8的对中仅一对可以具有每个r基团为氢(即,所述碳水化合物环可以在环中包括仅单个亚甲基(-ch2-))。
对于上面的化合物,所述糖苷配基如上所述。
在一个实施方案中,所述糖具有下式:
在另一个实施方案中,所述糖具有下式:
在某些实施方案中,所述化合物包括氨基(例如,-nh2)、羧酸基(-co2h)和磺酸基(例如,-oso3h),其随ph环境可以变成带电荷的基团(例如,-nh3+、-co2-、-oso3-)。应当理解,本发明的化合物包括它们的盐(例如,金属盐)。
如上面所指出的,本发明的化合物包括它们的重原子衍生物。所述重原子衍生物可用作内部标准品。在其它实施方案中,a型和b型糖苷配基具有一个或多个(例如,三个或更多个)被氘替换的氢原子或者一个或多个(例如,三个或更多个)被碳-13替换的碳原子,使得糖苷配基的质量增加一个或多个道尔顿。所述酶产物和内部标准品在质量上不同且质量的差异可以通过使用额外的原子(例如,使化合物的一部分的长度改变一个或多个亚甲基)或通过掺入重原子(例如,氘替代氢、13c替代碳、15n替代氮)来实现。
所述mps-ii酶产物和内部标准品在质量上不同且质量的差异可以通过使用额外的原子(例如,使化合物的一部分的长度改变一个或多个亚甲基)或通过掺入重原子(例如,氘替代氢、13c替代碳、15n替代氮)来实现。
代表性mps-ii试剂包括下述化合物。
在某些实施方案中,mps-ii底物具有下式:
其中l2、l3、r1、r2和n如上面关于式(iii)所述。在某些实施方案中,mps-ii底物具有下式:
其中l2、l3和r1如上面关于式(iii)所述。
代表性mps-ii底物具有下式:
从以上底物(mps-ii-s)形成的mps-ii产物具有下式:
其中l2、l3、r1、r2和n如上面关于式(iii)所述。
代表性mps-ii产物具有下式:
可用于测定从以上底物(mps-ii-s)形成的产物的mps-ii内部标准品具有下式:
其中l2、l3、r1、r2和n如上面关于式(iii)所述,且其中mps-ii-is的质量不同于mps-ii-p的质量,使得二者可通过质谱法辨别。如上面所指出的,mps-ii-is可以包括一个或多个重原子同位素(在上面的结构中未显示),或者可以具有结构变异(例如,底物的l2、l3、r1或r2中的一个或多个不同于内部标准品的l2、l3、r1或r2)。
代表性mps-ii内部标准品具有下式:
使用代表性mps-ii内部标准品可以测定从代表性mps-ii底物衍生出的代表性mps-ii产物。
mps-iiia试剂
在另一个实施方案中,本发明提供了由下式定义的mps-iiia试剂(s、p和is试剂)、它的盐及其重原子衍生物:
r1=糖苷配基,r2=h
或者
r1=h,r2=糖苷配基
r3=h、oh、nh2、nhso3h、oso3h且r4=h
或者
r4=h、oh、nh2、nhso3h、oso3h且r3=h
r5=h、oh、nh2且r6=h
或者
r6=h、oh、nh2且r5=h
r7=h、oh、nh2且r8=h
或者
r8=h、oh、nh2且r7=h
r9=ch2oh、ch2nh2且r10=h
或者
r10=ch2oh、ch2nh2且r9=h,
其中所述糖苷配基如上所述,且
前提条件是,r3和r4、r5和r6、以及r7和r8的对中仅一对可以具有每个r基团作为氢(即,所述碳水化合物环可以在环中包括仅单个亚甲基(-ch2-))。
在一个实施方案中,所述糖具有下式:
在另一个实施方案中,所述糖具有下式:
在某些实施方案中,所述化合物包括氨基(例如,-nh2)和酰氨基磺酸基(例如,-nhso3h),其随ph环境可以变成带电荷的基团(例如,-nh3+、-nhso3-)。应当理解,本发明的化合物包括它们的盐(例如,金属盐)。
所述mps-iiia酶产物和内部标准品在质量上不同且质量的差异可以通过使用额外的原子(例如,使化合物的一部分的长度改变一个或多个亚甲基)或通过掺入重原子(例如,氘替代氢、13c替代碳、15n替代氮)来实现。
代表性mps-iiia试剂包括下述化合物。
在某些实施方案中,mps-iiia底物具有下式:
其中l2、l3、r1、r2和n如上面关于式(iii)所述。
在某些实施方案中,mps-iiia底物具有下式:
其中l2、l3和r1如上面关于式(iii)所述。
代表性mps-iiia底物具有下式:
从以上底物(mps-iiia-s)形成的mps-iiia产物具有下式:
其中l2、l3、r1、r2和n如上面关于式(iii)所述。
代表性mps-iiia产物具有下式:
可用于测定从以上底物(mps-iiia-s)形成的产物的mps-iiia内部标准品具有下式:
其中l2、l3、r1、r2和n如上面关于式(iii)所述,且其中mps-iiia-is的质量不同于mps-iiia-p的质量,使得二者可通过质谱法辨别。如上面所指出的,mps-iiia-is可以包括一个或多个重原子同位素(在上面的结构中未显示),或者可以具有结构变异(例如,底物的l2、l3、r1、r2中的一个或多个不同于内部标准品的l2、l3、r1、r2)。
代表性mps-iiia内部标准品具有下式:
使用代表性mps-iiia内部标准品可以测定从代表性mps-iiia底物衍生出的代表性mps-iiia产物。
mps-iiib试剂
在另一个实施方案中,本发明提供了由下式定义的mps-iiib试剂(s、p和is试剂)、它的盐及其重原子衍生物:
r1=糖苷配基,r2=h
或者
r1=h,r2=糖苷配基
r3=h、oh、nh2、nhr11,其中r11=甲酰基、乙酰基、c=o((ch2)nch3),(n=1-6)且r4=h
或者
r4=h、oh、nh2、nhr11,其中r11=甲酰基、乙酰基、c=o((ch2)nch3),(n=1-6)且r3=h
r5=h、oh、nh2且r6=h
或者
r6=h、oh、nh2且r5=h
r7=h、oh、nh2且r8=h
或者
r8=h、oh、nh2且r7=h
r9=ch2oh、ch2nh2且r10=h
或者
r10=ch2oh、ch2nh2且r10=h,
其中所述糖苷配基如上所述,且
前提条件是,r3和r4、r5和r6、以及r7和r8的对中仅一对可以具有每个r基团作为氢(即,所述碳水化合物环可以在环中包括仅单个亚甲基(-ch2-))。
在上面定义的mps-iiib试剂的某些实施方案中,所述碳水化合物部分被氢原子替换;在该情况下,将氢原子添加至糖苷配基。这些试剂是mps-iiib酶产物和内部标准品的代表。
在一个实施方案中,所述糖具有下式:
在上式中,“nhac”表示“nh-c(=o)ch3”。
在某些实施方案中,所述化合物包括氨基(例如,-nh2),其取决于ph环境可以变成带电荷的基团(例如,-nh3+)。应当理解,本发明的化合物包括它们的盐(例如,金属盐)。
所述mps-iiib酶产物和内部标准品在质量上不同且质量的差异可以通过使用额外的原子(例如,使化合物的一部分的长度改变一个或多个亚甲基)或通过掺入重原子(例如,氘替代氢、13c替代碳、15n替代氮)来实现。
代表性mps-iiib试剂包括下述化合物。
在某些实施方案中,mps-iiib底物具有下式:
其中l2、l3、r1、r2、r3和n如上面关于式(iii)所述。
在某些实施方案中,mps-iiib底物具有下式:
其中l2、l3、r1和r3如上面关于式(iii)所述。
代表性mps-iiib底物具有下式:
从以上底物(mps-iiib-s)形成的mps-iiib产物具有下式:
其中l2、l3、r1、r2、r3和n如上面关于式(iii)所述。
代表性mps-iiib产物具有下式:
可用于测定从以上底物(mps-iiib-s)形成的产物的mps-iiib内部标准品具有下式:
其中l2、l3、r1、r2、r3和n如上面关于式(iii)所述,且其中mps-iiib-is的质量不同于mps-iiib-p的质量,使得二者可通过质谱法辨别。如上面所指出的,mps-iiib-is可以包括一个或多个重原子同位素(在上面的结构中未显示),或者可以具有结构变异(例如,底物的l2、l3、r1、r2或r3中的一个或多个不同于内部标准品的l2、l3、r1、r2或r3)。
代表性mps-iiib内部标准品具有下式:
使用代表性mps-iiib内部标准品可以测定从代表性mps-iiib底物衍生出的代表性mps-iiib产物。
mps-iva试剂
在另一个实施方案中,本发明提供了由下式定义的mps-iva试剂(s、p和is试剂)、它的盐及其重原子衍生物:
r1=糖苷配基,r2=h
或者
r1=h,r2=糖苷配基
r3=h、oh、nh2、nhr11,其中r11=甲酰基、乙酰基、c=o((ch2)nch3),(n=1-6)且r4=h
或者
r4=h、oh、nh2、nhr11,其中r11=甲酰基、乙酰基、c=o((ch2)nch3),(n=1-6)且r3=h
r5=h、oh、nh2且r6=h
或者
r6=h、oh、nh2且r5=h
r7=h、oh、nh2且r8=h
或者
r8=h、oh、nh2且r7=h
r9=ch2oh、ch2oso3h、ch2nh2、ch2nhso3h且r10=h
或者
r10=ch2oh、ch2oso3h、ch2nh2、ch2nhso3h且r9=h,
其中所述糖苷配基如上所述,且
前提条件是,r3和r4、r5和r6、以及r7和r8的对中仅一对可以具有每个r基团作为氢(即,所述碳水化合物环可以在环中包括仅单个亚甲基(-ch2-))。
在一个实施方案中,所述糖具有下式:
在某些实施方案中,所述化合物包括氨基(例如,-nh2)和磺酸基(例如,-oso3h),其随ph环境可以变成带电荷的基团(例如,-nh3+、-oso3-)。应当理解,本发明的化合物包括它们的盐(例如,金属盐)。
所述mps-iva酶产物和内部标准品在质量上不同且质量的差异可以通过使用额外的原子(例如,使化合物的一部分的长度改变一个或多个亚甲基)或通过掺入重原子(例如,氘替代氢、13c替代碳、15n替代氮)来实现。
代表性mps-iva试剂包括下述化合物。
在某些实施方案中,mps-iva底物具有下式:
其中l2、l3、r1、r2和n如上面关于式(iii)所述。r4在每次出现时独立地选自c1-c6烷基(例如,甲基)且m是0、1、2、3、4或5。
在其它实施方案中,mps-iva底物具有下式:
其中l2、l3、r1、r2、r3和n如上面关于式(iii)所述。
在某些实施方案中,mps-iva底物具有下式:
其中l2、l3、r1、r4和m如上面关于式(iv)所述。
在其它实施方案中,mps-iva底物具有下式:
其中l2、l3、r1和r3如上面关于式(iii)所述。
代表性mps-iva底物具有下式:
另一种代表性mps-iva底物具有下式:
从以上底物(mps-iva-s1)形成的mps-iva产物具有下式:
其中l2、l3、r1、r2、r4、n和m如上面在式(iv)中所述。
在另一个实施方案中,从以上底物(mps-iva-s2)形成的mps-iva产物具有下式:
其中l2、l3、r1和r3如上面关于式(iii)所述。
代表性mps-iva产物具有下式:
另一种代表性mps-iva产物具有下式:
可用于测定从以上底物(mps-iva-s1和mps-iva-s2)形成的产物的mps-iva内部标准品具有下式:
其中l2、l3、r1、r2、r3、r4、n和m如上所述,且mps-iva-is1和mps-iva-is2的质量分别不同于mps-iva-p1和mps-iva-p2的质量,使得二者可通过质谱法辨别。如上面所指出的,mps-iva-is1和mps-iva-is2可以包括一个或多个重原子同位素(在上面的结构中未显示),或者可以具有结构变异(例如,底物的l2、l3、r1、r2、r3或r4中的一个或多个不同于内部标准品的l2、l3、r1、r2、r3或r4)。
代表性mps-iva内部标准品具有下式:
另一种代表性mps-iva内部标准品具有下式:
使用代表性mps-iva内部标准品可以测定从代表性mps-iva底物衍生出的代表性mps-iva产物。
下面描述了另一个代表性mps-iva试剂集合。
另一种代表性mps-iva底物具有下式:
另一种代表性mps-iva产物标准品具有下式:
另一种代表性mps-iva内部标准品具有下式:
在上式中,x选自氟、甲基和甲氧基。
mps-vi试剂
在另一个实施方案中,本发明提供了由下式定义的mps-vi试剂(s、p和is试剂)、它的盐及其重原子衍生物:
r1=糖苷配基,r2=h
或者
r1=h,r2=糖苷配基
r3=h、oh、nh2、nhr11,其中r11=甲酰基、乙酰基、c=o((ch2)nch3),(n=1-6)且r4=h
或者
r4=h、oh、nh2、nhr11,其中r11=甲酰基、乙酰基、c=o((ch2)nch3),(n=1-6)且r3=h
r5=h、oh、nh2且r6=h
或者
r6=h、oh、nh2且r5=h
r7=ch2oh、ch2oso3h、ch2nh2、ch2nhso3h且r8=h
或者
r8=ch2oh、ch2oso3h、ch2nh2、ch2nhso3h且r7=h
r9=ch2oh、ch2nh2且r10=h
或者
r10=ch2oh、ch2nh2且r9=h,
其中所述糖苷配基如上所述,且
前提条件是,r3和r4、r5和r6、以及r7和r8的对中仅一对可以具有每个r基团作为氢(即,所述碳水化合物环可以在环中包括仅单个亚甲基(-ch2-))。
在一个实施方案中,所述糖具有下式:
在另一个实施方案中,所述糖具有下式:
在上式中,“acnh”表示“ch3c(=o)nh”。
在某些实施方案中,所述化合物包括氨基(例如,-nh2)和磺酸基(例如,-oso3h),其随ph环境可以变成带电荷的基团(例如,-nh3+、-oso3-)。应当理解,本发明的化合物包括它们的盐(例如,金属盐)。
所述mps-vi酶产物和内部标准品在质量上不同且质量的差异可以通过使用额外的原子(例如,使化合物的一部分的长度改变一个或多个亚甲基)或通过掺入重原子(例如,氘替代氢、13c替代碳、15n替代氮)来实现。
代表性mps-vi试剂包括下述化合物。
在某些实施方案中,mps-vi底物具有下式:
其中l2、l3、r1、r2和n如上面关于式(iii)所述。
在某些实施方案中,mps-vi底物具有下式:
其中l2、l3和r1如上面关于式(iii)所述。
代表性mps-vi底物具有下式:
从以上底物(mps-vi-s)形成的mps-vi产物具有下式:
其中l2、l3、r1、r2和n如上面关于式(iii)所述。
代表性mps-vi产物具有下式:
可用于测定从以上底物(mps-vi-s)形成的产物的mps-vi内部标准品具有下式:
其中l2、l3、r1、r2和n如上面关于式(iii)所述,且其中mps-vi-is的质量不同于mps-vi-p的质量,使得二者可通过质谱法辨别。如上面所指出的,mps-vi-is可以包括一个或多个重原子同位素(在上面的结构中未显示),或者可以具有结构变异(例如,底物的l2、l3、r1或r2中的一个或多个不同于内部标准品的l2、l3、r1或r2)。
代表性mps-vi内部标准品具有下式:
使用代表性mps-vi内部标准品可以测定从代表性mps-vi底物衍生出的代表性mps-vi产物。
mps-vii试剂
在一个实施方案中,本发明提供了由下式定义的mps-vii试剂(s、p和is试剂)、它的盐及其重原子衍生物:
其中
r1=糖苷配基,r2=h
或者
r1=h,r2=糖苷配基
r3=h、oh、nh2且r4=h
或者
r4=h、oh、nh2且r3=h
r5=h、oh、nh2且r6=h
或者
r6=h、oh、nh2且r5=h
r7=h、oh、nh2且r8=h
或者
r8=h、oh、nh2且r7=h
r9=cooh且r10=h
或者
r10=cooh且r9=h
前提条件是,r3和r4、r5和r6、以及r7和r8的对中仅一对可以具有每个r基团作为氢(即,所述碳水化合物环可以在环中包括仅单个亚甲基(-ch2-))。
对于上面的化合物,所述糖苷配基如上所述。
在上面定义的mps-vii试剂的某些实施方案中,所述碳水化合物部分被氢原子替换;在该情况下,将氢原子添加至糖苷配基。这些试剂是mps-vii酶产物和内部标准品的代表。
在一个实施方案中,所述糖具有下式:
在某些实施方案中,所述化合物包括氨基(例如,-nh2)和羧酸基(-co2h),其随ph环境可以变成带电荷的基团(例如,-nh3+或-co2-)。应当理解,本发明的化合物包括它们的盐(例如,金属盐)。
如上面所指出的,本发明的化合物包括它们的重原子衍生物。所述重原子衍生物可用作内部标准品。在某些实施方案中,a型和b型糖苷配基具有一个或多个(例如,三个或多个)被氘替换的氢原子或者一个或多个(例如,三个或多个)被碳-13替换的碳原子,使得糖苷配基的质量增加一个或多个道尔顿。所述酶产物和内部标准品在质量上不同且质量的差异可以通过使用额外的原子(例如,使化合物的一部分的长度改变一个或多个亚甲基)或通过掺入重原子(例如,氘替代氢、13c替代碳、15n替代氮)来实现。
在上面定义的mps-vii试剂的某些实施方案中,所述碳水化合物部分被氢原子替换;在该情况下,将氢原子添加至糖苷配基。这些试剂是mps-vii酶产物和内部标准品的代表。
代表性mps-vii试剂包括下述化合物。
在某些实施方案中,mps-vii底物具有下式:
其中l2、l3、r1、r2、r3和n如上面关于式(iii)所述。
在某些实施方案中,mps-vii底物具有下式:
其中l2、l3、r1和r3如上面关于式(iii)所述。
代表性mps-vii底物具有下式:
从以上底物(mps-vii-s)形成的mps-vii产物具有下式:
其中l2、l3、r1、r2、r3和n如上面关于式(iii)所述。
代表性mps-vii产物具有下式:
可用于测定从以上底物(mps-vii-s)形成的产物的mps-vii内部标准品具有下式:
其中l2、l3、r1、r2、r3和n如上面关于式(iii)所述,且其中mps-vii-is的质量不同于mps-vii-p的质量,使得二者可通过质谱法辨别。如上面所指出的,mps-vii-is可以包括一个或多个重原子同位素(在上面的结构中未显示),或者可以具有结构变异(例如,底物的l2、l3、r1、r2或r3中的一个或多个不同于内部标准品的l2、l3、r1、r2或r3)。
代表性mps-vii内部标准品具有下式:
使用代表性mps-vii内部标准品可以测定从代表性mps-vii底物衍生出的代表性mps-vii产物。
试剂盒
本发明的试剂可以有利地组合成试剂盒以执行酶测定。用于特定测定的试剂盒包括适当的酶底物和内部标准品对(例如,mps-ii-s和mps-ii-is)。在某些实施方案中,所述试剂盒包括超过一个底物/内部标准品对且可以用于测定超过一种酶(即,多路测定,其中可以在单次筛选中测定2种、3种、4种、5种或6种酶)。在其它实施方案中,所述试剂盒进一步包括用于执行测定的缓冲液。在其它实施方案中,所述试剂盒进一步包括酶产物,其可以用于调节质谱仪。在其它实施方案中,所述试剂盒进一步包括质量控制干燥血斑点。在试剂盒中还可以包括用于执行测定的说明书。
酶测定
本发明的试剂可以有利地用于测定与溶酶体贮积病有关的酶。在测定中,将一种或多种底物(s)与合适的酶来源(诸如来自新生儿筛查卡或尿样品的干燥血斑点)一起在合适的缓冲液中温育足够的时间以形成一种或多种产物(p),随后通过串联质谱法检测所述产物。所述测定也利用内部标准品(is),其在某些实施方案中与酶形成的产物在化学上相同,但是具有不同的质量(例如,重同位素取代的,诸如氘和/或碳-13取代)。所述温育在合适的缓冲液中完成以允许酶反应进行(例如,含有5mm乙酸钡和7.5mm乙酸铈的100mm甲酸铵ph4.5)。
有利地用本发明的试剂测定的酶包括以下的:
(a)α-l-艾杜糖醛酸酶,其作用于mps-i的底物以产生mps-i产物,且所述测定利用mps-i内部标准品;
(b)艾杜糖醛酸-2-硫酸酯酶,其作用于mps-ii的底物以产生mps-ii产物,且所述测定利用mps-ii内部标准品;
(c)类肝素n-硫酸酯酶,其作用于mps-iiia的底物以产生mps-iiia产物,且所述测定利用mps-iiia内部标准品;
(d)n-乙酰基-α-d-氨基葡糖苷酶,其作用于mps-iiib的底物以产生mps-iiib产物,且所述测定利用mps-iiib内部标准品;
(e)n-乙酰基半乳糖胺-6-硫酸酯-硫酸酯酶,其作用于mps-iva的底物以产生mps-iva产物,且所述测定利用mps-iva内部标准品;
(f)n-乙酰基半乳糖胺-4-硫酸酯-硫酸酯酶,其作用于mps-vi的底物以产生mps-vi产物,且所述测定利用mps-vi内部标准品;和
(g)β-葡糖醛酸糖苷酶,其作用于mps-vii的底物以产生mps-vii产物,且所述测定利用mps-vii内部标准品。
用于测定上述酶的代表性方法描述在wo2009/026252(pct/us2008/073516)、wo2010/081163(pct/us2010/020801)、wo2012/027612(pct/us2011/049224)和wo2013/070953(pct/us2012/064205)中,每篇明确地通过引用整体并入本文。本发明的试剂可以有利地用在这些方法中。
在实施例1-11中描述了使用本发明的mps-i、ii、iiia、iiib、iva和vi试剂的代表性测定。
本发明的测定可以包括没有脱离本发明的变型。下面描述了几种变型。
在第一个实施方案中,将底物和内部标准品与酶来源一起在测定缓冲液中温育,随后猝灭(例如,加入乙腈),然后质谱分析(例如,lc/msms)和定量糖-糖苷配基产物和内部标准品(应当指出,对于mps-i、mps-iiib和mps-vii,所述产物是糖苷配基(具有添加的氢))。
在第二个实施方案中,测定如在第一个实施方案中所述,但是用适合于萃取产物和内部标准品的有机溶剂(例如,乙酸乙酯)萃取酶反应混合物(任选地没有猝灭),将萃取的混合物浓缩至干燥,然后溶解在适合于流动注射质谱分析(例如,fia/msms)的溶剂中。
在第三个实施方案中,测定如在第二个实施方案中所述,但是在猝灭过程中加入阴离子交换树脂的悬浮液以捕集底物。
在第四个实施方案中,测定如在第一个实施方案中所述,但是在测定混合液(cocktail)(底物和内部标准品)中加入第二种酶(例如,糖原水解酶,诸如细菌β-n-乙酰基半乳糖胺酶),其适合于切割最初的硫酸酯酶产物(除去了硫酸酯的糖-糖苷配基),但是不会切割底物。在萃取、浓缩和重溶以后,进行质谱分析(例如,fia/msms),从而导致糖苷配基产物和内部标准品的定量。在某些情况下,可能存在在干燥血斑点样品中的内源性酶,其可以作用于硫酸化的糖-糖苷配基底物以切割糖苷键(即人己糖胺酶a)。在该情况下,可以加入该内源性酶的抑制剂来阻断内源性酶在添加的底物上的作用。选择该抑制剂使得不会阻断向测定中添加的糖原水解酶的作用。
在第五个实施方案中,测定如在第二个实施方案中所述,但是在测定混合液(底物和内部标准品)中加入适合于切割选择性的硫酸酯酶糖-糖苷配基底物的第二种酶(例如,糖原水解酶)。猝灭以后,使用质谱分析(例如,lc/msms)来定量糖苷配基产物和内部标准品。在该实施方案的改进中,还将内源性活性的抑制剂(例如,人己糖胺酶a)加入测定混合液中。
在第六个实施方案中,将底物和内部标准品与酶来源一起在测定缓冲液中温育,然后加入缓冲液以转换ph(例如,至ph6)以优化第二种酶(例如,糖原水解酶)的活性,随后加入糖原水解酶(例如,细菌β-n-乙酰基半乳糖胺酶)和温育(例如,1-2小时)。然后将样品猝灭,并使用质谱分析(例如,lc/msms)定量糖苷配基产物和内部标准品。在该实施方案的改进中,还将内源性酶活性的抑制剂(例如,人己糖胺酶a)加入测定混合液中。
在第七个实施方案中,测定如在第六个实施方案中所述,但是用适合于萃取产物和内部标准品的有机溶剂(例如,乙酸乙酯)萃取酶反应混合物(任选地没有猝灭),将萃取的混合物浓缩至干燥,然后溶解在适合于流动注射分析(例如,fia/msms)的溶剂中。在该实施方案的改进中,还将内源性酶活性的抑制剂(例如,人己糖胺酶a)加入测定混合液中。
在第八个实施方案中,对于上面的利用有机溶剂萃取来分离产物和内部标准品的实施方案,在除去溶剂以后,加入合适的酰化剂(例如乙酸酐)和合适的碱(例如,三乙胺)在合适的溶剂中的溶液,并将得到的组合温育(1-2小时)以提供酰化的(例如,乙酰化的)糖苷配基产物和内部标准品,其在ms分析中具有增加的灵敏度。
在第九个实施方案中,测定如在第八个实施方案中所述,但是将酰化剂和碱包含在萃取(例如,乙酸乙酯)溶剂中以造成糖苷配基和内部标准品在萃取过程中或在将萃取物温育(例如,1-2小时)以后变得酰化(例如,乙酰化)。
在第十个实施方案中,将底物和第二种酶(糖原水解酶)与酶来源一起在测定缓冲液中温育,随后猝灭,然后荧光分析以定量荧光产物。在该实施方案的改进中,还将内源性酶活性的抑制剂(例如,人己糖胺酶a)加入测定混合液中。对于该实施方案,使用具有a型糖苷配基的底物。
在第十一个实施方案中,将底物与酶来源一起在测定缓冲液中温育,然后加入缓冲液以转换ph(例如,至ph6)以优化第二种酶(例如,糖原水解酶)的活性,随后加入糖原水解酶(例如,细菌β-n-乙酰基半乳糖胺酶)和温育(例如,1-2小时),然后猝灭和荧光产物的荧光分析定量。在该实施方案的改进中,还将内源性酶活性的抑制剂(例如,人己糖胺酶a)加入测定混合液中。对于该实施方案,使用具有a型糖苷配基的底物。
在某些实施方案中,在本发明的方法中还包括额外的测定选择。下面描述了两种选择。
测定选择1.将期望的底物集合与酶来源一起在合适的缓冲液中温育以后,用合适的溶剂诸如乙酸乙酯对反应物进行液-液萃取。如果在不使用第二种酶(糖原水解酶)来产生糖苷配基的情况下测定mps-ii,应当用合适的酸诸如柠檬酸将混合物酸化至ph约2-3,使得mps-ii-p的羧酸酯基(carboxylategroup)将被质子化和更好地萃取进乙酸乙酯中。如果在该测定中使用第二种酶来除去糖,糖苷配基将在没有酸化下较好地萃取进溶剂中,因为它不具有羧基。液-液萃取步骤的目的是双重的:(1)萃取导致大部分缓冲盐的除去,所述缓冲盐会干扰质谱仪中的电离过程;和(2)萃取导致大部分酶产物的萃取和酶底物的微小萃取。这是有用的,因为底物可以通过质谱仪的电离源中硫酸盐的损失而部分地分解以形成产物,并且人们仅希望定量酶促地产生的产物。液-液萃取以后,将乙酸乙酯层转移至新容器,并通过蒸发除去溶剂。将残余物溶解在适合于注射进质谱仪中的溶剂中。一种例子溶剂是水性的甲酸铵/甲醇混合物。以多重反应监测模式检测产物和内部标准品,其中将前体离子分离在第一个四极中,然后进行碰撞诱导的解离以形成一种或多种产物离子。将一种这样的产物离子分离在第三个四极中并通过离子检测器(串联质谱法)检测。以工作循环方式单独地监测每个片段化反应(每种产物和内部标准品用一个片段化反应),从而定量全套产物和内部标准品。为了得到产物的摩尔数,将产物的质谱法信号(离子计数)除以内部标准品的质谱法信号,并将该比率乘以加入测定中的内部标准品的摩尔数。
测定选择2.以上测定的一种变体利用改进的质谱法前(pre-massspectrometry)样品后处理(workup)。在允许从底物酶促地产生产物的温育以后,将合适的阴离子交换树脂的小等分试样加入混合物中。一种实例树脂是来自whatmann的de52。众所周知,阴离子通过与阴离子交换树脂上的阳离子的静电相互作用而结合,在该情况下,所有阴离子分析物将结合至树脂。底物mps-i-s、mps-ii-s、mps-iiia-s、mps-iva-s、mps-vi-s和mps-vii-s含有羧酸酯(mps-i-s和mps-vii-s)或硫酸酯,且因而将结合至所述树脂。mps-i-p、mps-i-is、mps-iva-p、mps-iva-is、mps-vi-p、mps-vi-is、mps-vii-p和mps-vii-is缺乏电荷或含有正电荷(mps-iiia-p和mps-iiia-is),且因而不会结合至所述树脂。mps-iiib-s、mps-iiib-p和mps-iiib-is也缺乏负电荷且不会结合至所述树脂。mps-ii-s、mps-ii-p和mps-ii-is都是阴离子的,且因而都会结合至所述树脂。在该测定选择中,测定缓冲液含有重组α-l-艾杜糖醛酸酶,其作用于mps-ii-p和mps-ii-is,不作用于mps-ii-s,以从糖苷配基除去艾杜糖醛酸残基,从而剩下游离的缺少电荷的糖苷配基。因而,得到的从mps-ii-p和mps-ii-is衍生出的分析物不会结合至阴离子交换树脂。如果包括重组α-l-艾杜糖醛酸酶,测定不可以包括mps-i-s,因为所述酶将作用于mps-i-s以产生mps-i-p。α-l-艾杜糖醛酸酶的应用不限于其中加入阴离子交换树脂的那些mps-ii测定。对于其中没有使用阴离子交换剂的所有其它mps-ii测定,也可以进行该酶的添加。
加入阴离子交换树脂以后,如在测定选择1中一样用乙酸乙酯萃取混合物,并且所有未结合至树脂的分析物将被萃取进乙酸乙酯中。然后如在测定选择1中所述处理乙酸乙酯层用于串联质谱法分析。
多路测定.本发明的方法提供了mps-i、mps-ii、mps-iiia、mps-iiib、mps-iva、mps-vi和mps-vii中的一种或多种(包括它们的任意组合)的分析。对于利用质谱法来定量测定产物的实施方案,在某些实施方案中,用于每个测定的产物是质量不同的。每种产物的质量不同,使得单个测定可以用于定量所有测定产物。通过底物的选择来实现质量差别性。
上述的mps-i、mps-ii、mps-iiia、mps-iiib、mps-iva、mps-vi和mps-vii的代表性底物会提供质量不同的产物(即,没有两种产物具有相同的质量)。与它们的对应内部标准品(参见上述的mps-i、mps-ii、mps-iiia、mps-iiib、mps-iva、mps-vi和mps-vii的代表性内部标准品)一起,结果是一次执行和分析超过单个测定的能力。
在其它实施方案中,超过一种产物离子可以具有相同的质量。在这些实施方案中,可以得到定量,只要从这些等比重产物衍生出的片段质量是不同的(即,母离子质量/片段离子质量的组合对于混合物中要定量的每种物质而言是独特的)。
在一个方面,本发明提供了使用本文描述的试剂同时测定mps-i、mps-ii、mps-iiia、mps-iiib、mps-iva、mps-vi和mps-vii或其任何亚类的方法。
硫酸酯酶测定:mpsii、iiia、iva和vi筛查
在另一个方面,本发明提供了用于新生儿筛查的、用于检测与溶酶体贮积病有关的硫酸酯酶(mpsii、iiia、iva和vi)的测定、试剂和试剂盒。
使用串联质谱法的溶酶体酶测定可用于溶酶体贮积病的新生儿筛查。所述测定利用以下通式结构的底物:硫酸酯-糖-糖苷配基。这些底物被溶酶体硫酸酯酶作用以产生缺乏硫酸酯的产物:糖-糖苷配基。在某些测定中,使用串联质谱法检测糖-糖苷配基产物。
本发明提供了那些硫酸酯酶测定的替代测定。在本发明的测定的某些方面,将第二种酶加入测定混合液中从而实现糖的除去以提供糖苷配基。糖苷配基的串联质谱法检测比糖-糖苷配基的检测更灵敏。除去糖的第二种酶不会作用于硫酸化的糖(即,所述第二种酶不会作用于硫酸酯酶的底物)。因而,本发明提供了一种包括以下额外步骤的方法:将合适的糖原水解酶或合适的裂解酶加入测定混合液中以产生糖苷配基作为最终的酶产物,其然后通过串联质谱法来检测。
本文中使用的术语“糖原水解酶”表示水解糖苷的酶。术语“裂解酶”表示从糖c2除去质子并消除糖苷氧(例如,糖苷配基离去基团)以提供不饱和的糖衍生物的酶。
合适的第二种酶(例如,糖原水解酶和裂解酶)的特征在于,它们不会从本文描述的酶产物的糖苷配基切割糖(例如,它们仅仅在除去硫酸酯以后切割糖),它们不会作用于酶底物本身,并且它们不会被在本发明的测定中加入的抑制剂(为了阻断存在于干燥血斑点中的内源性酶的作用,如己糖胺酶a,其可以切割硫酸化的底物以提供糖苷配基)抑制。
合适的第二种酶包括糖原水解酶和裂解酶。
代表性的糖原水解酶包括人己糖胺酶a、细菌n-乙酰基己糖胺酶、细菌β-n-乙酰基半乳糖胺酶(例如,类芽孢杆菌属种ts12)、α-l-艾杜糖醛酸酶、β-半乳糖苷酶(曲霉菌属)和α-葡萄糖苷酶(酵母)。
代表性的裂解酶包括肝素裂解酶(肝素酶)和类肝素酶。
测定方法.在一个方面,本发明提供了用于筛查mpsii、iiia、iva和vi的方法。所述方法测定特定酶,所述酶的缺乏会导致溶酶体贮积病状况。所述方法有利地测定艾杜糖醛酸-2-硫酸酯酶(mps-ii)、类肝素n-硫酸酯酶(mps-iiia)、n-乙酰基半乳糖胺-6-硫酸酯-硫酸酯酶(mps-iva)和n-乙酰基半乳糖胺-4-硫酸酯-硫酸酯酶(mps-vi)中的一种或多种。
如上面所指出的,在本发明的方法中,利用第二种酶来提高质谱测定实施方案的灵敏度和产生在荧光测定实施方案中的荧光团。根据所述方法,使合适的硫酸酯酶底物(即,硫酸酯-糖-糖苷配基)与要评估硫酸酯酶活性的样品接触。当所述样品包括硫酸酯酶时,将所述底物酶促地转化成最初的酶产物(即,糖-糖苷配基)。在本发明的方法中,第二种酶(例如,糖原水解酶)作用于最初的酶产物以提供第二酶产物(即,糖苷配基)。与先前的测定(其中不存在第二种酶,且其依赖于最初形成的酶产物糖-糖苷配基的分析)相比,通过串联质谱法对第二酶产物(即,糖苷配基)的分析会提供增加的灵敏度。对于含有a型糖苷配基的底物,第二种酶仅在硫酸酯被除去以后起作用以释放荧光糖苷配基。这允许通过荧光分析来测定硫酸酯酶。
对于荧光测定而言,所述猝灭可以包括缓冲液来将ph升高至约10,使得糖苷配基的酚羟基被去质子化,从而赋予产物(糖苷配基)高荧光性。
在测定中,将一种或多种底物(s)与合适的酶来源(诸如来自新生儿筛查卡或尿样品的干燥血斑点)一起在合适的缓冲液中温育足够的时间以形成一种或多种产物(p1),随后使所述产物遇到第二种酶(即,糖原水解酶)以提供第二酶产物(p2),其通过串联质谱法进行检测。所述测定也利用内部标准品(is),其在某些实施方案中与酶形成的产物在化学上相同,但是具有不同的质量(例如,重同位素取代的,诸如氘和/或碳-13取代)。在某些测定中,所述内部标准品也被第二种酶起作用以形成最终的内部标准品,其通过串联质谱法进行检测。所述温育在合适的缓冲液中实现以允许酶反应进行。合适的缓冲液是例如含有7.5mm乙酸钡和5mm乙酸铈的100mm甲酸铵ph4.5。
有利地用本发明的试剂测定的酶包括以下的:
(a)艾杜糖醛酸-2-硫酸酯酶,其作用于mps-ii的底物以产生mps-ii产物,且所述测定利用mps-ii内部标准品;
(b)类肝素n-硫酸酯酶,其作用于mps-iiia的底物以产生mps-iiia产物,且所述测定利用mps-iiia内部标准品;
(c)n-乙酰基半乳糖胺-6-硫酸酯-硫酸酯酶,其作用于mps-iva的底物以产生mps-iva产物,且所述测定利用mps-iva内部标准品;和
(d)n-乙酰基半乳糖胺-4-硫酸酯-硫酸酯酶,其作用于mps-vi的底物以产生mps-vi产物,且所述测定利用mps-vi内部标准品。
在某些实施方案中,在本发明的方法内还包括额外的测定选择。上面指出的测定选择1和2可以用在这些测定方法中。
试剂.用于筛查mpsii、iiia、iva和vi的试剂包括用于筛查mpsii、iiia、iva和vi的底物(s)、产物(p)和内部标准品(is)。
所述试剂可以有利地用于测定酶。所述试剂包括酶底物(s)、酶产物(p)和测定内部标准品(is)。在某些实施方案中,将一种或多种底物(s)和它们的对应内部标准品(is)与合适的酶来源(诸如来自新生儿筛查卡或尿样品的干燥血斑点)一起在合适的缓冲液中温育足够的时间以形成一种或多种产物(p),随后使所述产物遇到第二种酶(例如,糖原水解酶)以提供第二酶产物,其通过串联质谱法进行检测。在某些实施方案中,所述内部标准品(is)与酶形成的产物在化学上相同,但是所述标准品具有不同的质量(例如,重同位素取代的,诸如氘和/或碳-13取代)。
在这些硫酸酯酶的测定中有用的试剂包括mps-ii、mps-iiia、mpsiva和mps-vi的底物(s)、产物(p)和内部标准品(is),包括本文描述的那些。
在实施例7-11中描述了mps-ii、mps-iiia、mpsiva和mps-vi的代表性测定。
mpsiva和vi筛查的代表性测定和结果
如上所述,使用糖原水解酶和裂解酶来提高使用质谱法测定硫酸酯酶的灵敏度和提供用于荧光测定的荧光糖苷配基产物。在一个有关的方面,提供了一种方法,其进一步包括添加抑制剂以阻断内源性糖原水解酶活性的步骤。
合适的抑制剂会阻断内源性糖原水解酶活性,但是不会显著地抑制添加的糖原水解酶的活性。干燥血斑点含有例如己糖胺酶a,其可以作用于硫酸化的底物以在一个步骤中形成糖苷配基。在该情况下,当通过串联质谱法或荧光测定法测量糖苷配基以便定量硫酸酯酶时,最佳地加入己糖胺酶的抑制剂。添加的抑制剂应当不会显著地抑制第二种酶,所述第二种酶加入测定中以将最初的硫酸酯酶产物转化成糖苷配基。
合适的抑制剂会阻断生物样品中的己糖胺酶,但是不会完全阻断第二种酶。所述抑制剂可能部分地阻断后者,但是不会那么完全地阻断,所以后者可以将大多数(如果不是全部的话)最初的硫酸酯酶产物转化成它的糖苷配基。合适的抑制剂会抑制人己糖胺酶(hexosaminidinase)a、人己糖胺酶b和/或人己糖胺酶x。
代表性的抑制剂包括(z)-o-(2-乙酰氨基-2-脱氧-d-吡喃葡萄糖亚基)-氨基n-苯基氨基甲酸酯(z-pug-nac)、1-脱氧野尻霉素、栗精胺、苦马豆素、打碗花精b2、isofagamine、达菲、葡糖酸肟基内酯、葡糖醛酸以及它的内酯和内酰胺、扎那米韦、米格列醇、苯乙基取代的葡萄糖-和半乳糖-咪唑、n-羟基乙基脱氢野尻霉素、galnac噻唑啉和glcnac噻唑啉。
下面描述了mps-iva和mps-vi的代表性测定。
粘多糖贮积症iva的代表性测定.在一个实施方案中,提供了mps-iva(也被称作a型莫基奥综合征(morquioasyndrome))的一种测定,以检测n-乙酰基半乳糖胺-6-硫酸酯酶(也被称作galns,并在本文中与该术语可互换地使用),即在mps-iva中缺乏的酶。galns底物是与糖苷配基连接的n-乙酰基-半乳糖-6-硫酸酯,其中r1是正丁基,l2是-ch2ch2-,l3是-(ch2)5-,且r3是苯基(参见下文)。所述测定包括添加酶,其仅在6-硫酸酯已经被除去以后从糖苷配基除去n-乙酰基半乳糖胺。从糖苷配基除去n-乙酰基半乳糖胺的酶是β-己糖胺酶(例如,人己糖胺酶a),其将β-糖苷切割成n-乙酰基半乳糖胺和n-乙酰基葡糖胺残基。所述生物样品可以含有人己糖胺酶a和其它人己糖胺酶。人己糖胺酶a可以作用于mps-iva底物以产生糖苷配基。如在表1中所示,人己糖胺酶a可以切割糖苷,即使在所述糖在6-位被硫酸化时。参见各行,其中z-pug-nac被省略。
表1也表明,干燥血斑点中的内源性己糖胺酶a的量会在galns的测定中造成问题,所述测定使用干燥血斑点用于a型莫基奥综合征的新生儿筛查。
表1.galns的测定结果.
在某些实施方案中,在测定中使用细菌酶,即,得自类芽孢杆菌属种ts12的β-n-乙酰基半乳糖胺酶(β-nga)(j.biol.chem.2011,286,14065-14072)。该酶在结构上与人己糖胺酶无关,且在表1中显示的数据表明,该细菌酶将β糖苷切割成n-乙酰基-半乳糖胺(当它没有在6-位硫酸化时),并且该酶不会显著地作用于所述底物(当它带有硫酸酯时)。
在某些实施方案中,在所述测定中使用人己糖胺酶a的抑制剂来阻断人己糖胺酶a对galns底物的作用。在某些实施方案中,所述抑制剂不会显著地抑制β-nga。
在以上测定中使用的galns底物具有以下结构:
代表性的测定使用在新生儿筛查卡上的干燥血斑点作为galns的来源。这些测定也使用β-nga以及人己糖胺酶a的抑制剂:z-pug-nac((z)-o-(2-乙酰氨基-2-脱氧-d-吡喃葡萄糖亚基)-氨基n-苯基氨基甲酸酯)。
干燥血斑点中的galns酶作用于上面显示的galns底物以释放产物(即,没有6-硫酸酯的galns底物)。测定混合液中的细菌β-nga作用于没有6-硫酸酯的galns底物以释放糖苷配基,且不会作用于galns底物。z-pug-nac向测定混合液中的添加会阻断内源性人己糖胺酶a作用于galns底物以产生糖苷配基的能力。然后通过串联质谱法检测所述糖苷配基。因而,糖苷配基形成标志着galns的作用,并通过检测糖苷配基(而不是最初形成的脱硫酸化产物)来获得灵敏度。
在实施例10中描述了mps-iva的代表性硫酸酯酶测定。
mps-vi的代表性测定.在另一个实施方案中,本发明提供了为mps-vi开发的一种测定,以检测芳基硫酸酯酶b(asb),即在mps-vi(也被称作maroteaux-lamy综合征)中缺乏的酶。
在所述测定中使用以下asb(mps-vi)底物:
asb从来自asb底物的n-乙酰基-半乳糖胺-4-硫酸酯基团除去4-硫酸酯。在本发明之前,不可得到关于人己糖胺酶a作用于n-乙酰基-半乳糖胺-4-硫酸酯的能力的数据。重组人己糖胺酶a作用于asb底物以释放糖苷配基。通过uhplc-ms/ms来检测糖苷配基(参见图1)。
图1表明,该糖苷配基的量随着加入混合物中的己糖胺酶a的量而增加。所述混合物包括在100mm甲酸铵缓冲液(ph5.6)中的1mmasb底物,所述缓冲液含有指定量的己糖胺酶。在37℃温育5小时以后,通过uhplc-ms/ms分析混合物。糖苷配基信号不是由于具有糖苷配基的底物的污染,因为当省略己糖胺酶a时没有看到糖苷配基。糖苷配基不是在质谱仪的电喷射电离源中的asb的切割的结果,因为糖苷配基的量随着己糖胺酶a的增加而增加(图1),并且糖苷配基的uhplc保留时间匹配真实糖苷配基的uhplc保留时间,这不同于asb底物的保留时间。因而,己糖胺酶a作用于asb底物以产生糖苷配基。因为己糖胺酶a作用于asb底物以产生糖苷配基,该酶不可用在asb的偶联测定中,此外重要的是抑制己糖胺酶a从而不会直接从asb底物产生糖苷配基。
已经确立了人己糖胺酶a对asb底物的作用,使用为galns开发的相同方案,即加入β-nga和z-pug-nac,开发了asb测定。结果给出在下面的表2中。如关于galns测定(参见实施例10)那样进行实验,但是用1mmasb底物替换galns底物。
表2.asb的测定结果.
实验1(含有血液的完整测定)与实验2(无血液对照)的对比表明,存在的β-nga在两个实验中将大多数asb内部标准品转化成它的糖苷配基。含有血液的糖苷配基的量(1,570,000)远高于在无血液对照中形成的量(58,300)。实验3具有血液和z-pug-nac,但是无β-nga,并将asb产物的量(371,000)与在有β-nga存在下它的糖苷配基的信号(1,570,000)进行对比,再次表明将产物转化成糖苷配基的灵敏度优点。最后,实验4不具有血液且无β-nga或z-pug-nac,表明asb底物被少量产物(3,340)和它的糖苷配基(33,800)污染。
本发明提供了具有下文描述的性能的任何酶用于执行galns和asb酶活性的测定的用途:
(a)仅在糖不带有在4-或6-位的硫酸酯时,在本文描述的测定中使用的酶将会切割所述糖以产生糖苷配基;和
(b)在本文描述的测定中使用的酶不会被加入测定混合物中的抑制剂显著地抑制,所述抑制剂显著地抑制存在于生物样品(例如,干燥血斑点)中的人己糖胺酶a,所述生物样品是要测定的溶酶体酶的来源。
荧光检测.在某些实施方案中,本发明的某些测定也可用在使用荧光方法的本文描述的酶的测定中。在一个方面,糖苷配基是发荧光的,但是当它与糖形成糖苷键时荧光减少。因而,在一个方面,芳基硫酸酯酶b(asb)和细菌n-乙酰基半乳糖胺酶的组合作用导致荧光信号的增加,这归因于高荧光糖苷配基的形成。下面显示了适合与本发明的测定一起使用的发荧光的糖苷。
上面显示的发荧光的糖苷是发荧光的asb底物,其含有添加至带有额外取代基(r基团)的7-羟基香豆素的糖苷。asb和随后细菌n-乙酰基半乳糖胺酶的连续作用产生被取代的7-羟基香豆素,其比糖苷发更多的荧光。因而,通过观察荧光信号的增加来测定asb。还可以使用其它荧光团,只要糖苷是asb和n-乙酰基半乳糖胺酶的底物,并且当糖苷被切割时荧光团改变它的荧光强度。应当理解,当苯酚由于去质子化变成苯酚盐时,荧光强度在高ph显著地增加。实验性地,在某些实施方案中,用ph10.5缓冲液猝灭测定以揭示荧光。
在以前,不存在该方法用于测定asb的用途或酶仅在糖苷没有被硫酸化时选择性地切割糖苷的用途。该方法还可以用于测定galns,其使用含有在糖残基的6-位的硫酸酯的合适底物。本发明提供了使用基于n-乙酰基-半乳糖胺-6-硫酸酯的底物的测定,所述底物与其它底物(诸如作为糖苷连接至半乳糖-6-硫酸酯的4-甲基伞形酮)相比更快地被galns作用。
为了例证而不是限制本发明的目的,提供了以下实施例。
具体实施方式
实施例1
代表性mps-i和mps-ii试剂的合成
在该实施例中,描述了代表性mps-i(mps-i-s-乙酰基-c6和mps-i-is-乙酰基-c6)试剂和mps-ii(mps-ii-s-戊酰基-c6和mps-ii-is-戊酰基-c6)试剂的合成。
1-f-2,3,4-三乙酰氧基-艾杜糖醛酸甲酯的合成
下面描述了1-f-2,3,4-三乙酰氧基-艾杜糖醛酸甲酯的合成。
步骤1
在0℃在氮气下将50.05g(133.0mmol)部分的1,2,3,4-四-o-乙酰基-α,β-吡喃葡萄糖醛酸甲酯(carbosynth,uk)悬浮于295ml33%hbr/乙酸(acroscat.123180010)中并在0℃搅拌15分钟。使反应物温热至室温并继续搅拌3小时。将反应物用295ml甲苯稀释,然后在旋转蒸发器上浓缩(30-35℃水浴,用水泵抽吸)。将残余物溶解在800mletoac中,用500ml冰冷的饱和nahco3水溶液洗涤,用冰冷的盐水洗涤,然后经无水na2so4干燥。将溶液过滤,并如上通过旋转蒸发除去溶剂。将化合物在高真空下在室温放置1小时。质子-nmr(cdcl3)与产物一致。使用20%etoac/己烷(用5%的h2so4在meoh中的溶液炭化(charring))在硅胶(silica)上的tlc显示在rf约0.6处的产物,起始原料在rf约0.5。刚好在起点以上看到两个小斑点。
将得自上面的粗制溴化物溶解在600ml无水乙腈中并在氮气下搅拌约5分钟以溶解所述溴化物。将烧瓶包裹在铝箔中以避光。一次性加入20.27gagf(oakwoodcat.002862)。将反应物在暗处在氮气下在室温搅拌24小时。使用20%etoac/己烷(用5%的h2so4在meoh中的溶液炭化)在硅胶上的tlc显示在rf约0.5处的产物,起始原料在rf约0.6。在抽吸下穿过硅藻土垫过滤混合物,然后通过旋转蒸发(30-35℃水浴,用水泵抽吸)浓缩滤液。将180g硅胶加载进玻璃柱,并使己烷类穿过柱。将化合物溶解在etoac中,并加入最小量的干燥硅胶。通过旋转蒸发除去etoac,并将得到的化合物/硅胶混合物加载到柱的顶部上。在低气压下运行柱。最初,使1l100%己烷类穿过柱且没有洗脱产物。然后用2l20%etoac/己烷类运行柱,且没有洗脱产物。1l25%etoac/己烷类也没有洗脱产物。最后用3l30%etoac/己烷类运行柱,并洗脱所有产物。将级分合并,并通过旋转蒸发除去溶剂以产生白色结晶固体。将产物在高真空下在室温干燥1小时以产生27.0g产物(60.4%收率)。在cdcl3中的质子-nmr与产物一致。
步骤2
将6.0g得自上面的氟化物和6.0gnbs(aldrichcat.b81255-500g,从热水重结晶,在高真空下干燥过夜)溶解在240mlccl4(jtbakercat.1512-3)中,并在置于紫外灯(450w,在石英夹套中的aceglass7825-34灯,用水循环来冷却灯)旁边的圆底烧瓶中在氮气下搅拌。将具有相同反应的另一个圆底烧瓶放在相同紫外灯的对侧。以该方式同时运行两个反应。
通风柜的外侧用铝箔覆盖以保护化学家。将灯开启并将反应物在78℃回流2小时。将灯关闭,并将约6g部分的nbs加入每种反应物中,将紫外灯再次开启,并允许反应物继续回流。另外2小时以后,如上加入额外的约6g部分的nbs,并在有紫外灯存在下继续回流另外3小时。共7小时以后,将灯关闭,并将反应物冷却至室温。将每个反应物穿过玻璃绒过滤,并将所述绒用50mlccl4洗涤。通过旋转蒸发(35℃水浴,用水泵抽吸)除去溶剂。使用30%etoac/己烷类的tlc显示在rf约0.6处的产物斑点,其运行到刚好起始原料上面(用5%的h2so4在meoh中的溶液炭化)。在含有180g硅胶的柱上同时纯化4×6g规模反应物。将化合物溶解在etoac中,加入最小量的干燥硅胶,通过旋转蒸发除去etoac,然后将硅胶/化合物混合物加载至柱的顶部。用1l100%己烷类运行柱,没有洗脱产物。用2l10%etoac/己烷类运行柱,没有洗脱产物。用3l20%etoac/己烷类运行柱,产物洗脱为通过tlc看到的两个斑点中的较慢者。抛弃含有次要产物(较快的tlc斑点)的级分(加入的3l的从700ml至1300ml)。将所有含有产物的级分(加入的3l的从1400ml至2500ml)合并,并通过旋转蒸发除去溶剂,产生粘稠的黄色液体,其在静置过夜后变成结晶固体。将该固体在高真空下放置12小时。从4×6g反应物得到18.90g总产物(63.3%收率)。
步骤3
将9.92g得自上面的溴化物溶解在154ml甲苯(非干燥;macronchemicals目录号4483-4l)中并在氮气下搅拌。加入10.0mlbu3snh(aldrich234188-10g或acros215730500),并将混合物在110℃回流1小时。tlc(10%的etoac在甲苯中的溶液)表明剩下一些起始原料,所以继续回流另外1小时。重复tlc,并指示反应结束。将圆底烧瓶冷却至室温,并通过旋转蒸发(约40℃水浴,用水泵抽吸)除去溶剂。
将180g硅胶加载进柱中。将粗残余物吸附到硅胶上并象前面一样加载到柱上。用1l100%甲苯运行柱,没有洗脱产物。用1l10%etoac/甲苯运行柱,没有洗脱产物。用2.5l20%etoac/甲苯运行柱,产物洗脱为在rf约0.6处的两个斑点中的较慢者(在10%etoac/甲苯中的tlc)。在rf约0.7处的较快斑点是葡糖醛酸,次要产物。抛弃含有较快tlc斑点的级分(2.5l的从600ml至1000ml)。将含有产物的级分(2.5l的从1100ml至2100ml)合并和通过旋转蒸发浓缩(约40℃水浴,用水泵抽吸),并在高真空下放置3小时以产生5.2g产物(64.7%收率)。产物在纯化后是澄清的、粘稠的液体。
糖苷配基制备
糖苷配基可以通过两种方法来制备。第一种方法利用市售的单-boc-1,6-己烷二胺向ho-ph-nhco-ch=ch2的迈克尔加成,随后是仲胺的乙酰化、boc的除去和伯胺的苯甲酰化。发生苯酚-oh的一些苯甲酰化,但是该酯在样品后处理之前通过皂化来切割。第二种方法包括用苯甲酰氯处理单-boc-1,6-己烷二胺,除去boc以产生单-苯甲酰基-1,6-己烷二胺,将其用在迈克尔加成中,随后将仲胺乙酰化。因为苯甲酰化和boc除去都是几乎定量的,每个路线是基本上等效的。
对称二胺的单-苯甲酰化(tang,w.;fang,s.tetrahedronlett.2008,49,6003)
向苯甲酸甲酯(1.0g,7.34mmol)中加入戊烷-1,5-二胺(0.75g,7.34mmol)和水(0.37ml),并将混合物在恒定搅拌下加热至100℃保持24小时。将反应混合物冷却至室温,并直接加载到短硅胶柱(在加载反应混合物之前,所述硅胶柱用4%的三乙胺在氯仿中的溶液预冲洗,随后用100%氯仿预冲洗)上。在用30%的甲醇在氯仿中的溶液洗脱后,得到作为淡黄色油的期望的单-苯甲酰基化的产物(0.80g,53%)。1hnmr(300mhz,meod)δ7.83(d,j=7.4hz,2h),7.58-7.31(m,4h),3.41(t,j=7.0hz,2h),2.76(t,j=7.2hz,2h),1.74-1.21(m,6h)。[m+h]+的ms(esi+);计算:207.1,实测:207.2。
方法1.将4-氨基苯酚(50g,458mmol)在ch2cl2(400ml)中的溶液和饱和nahco3水溶液(400ml)在室温搅拌10min,然后逐滴加入丙烯酰氯(40.9ml,503.8mmol),并将反应物在室温搅拌另外6小时。将得到的固体通过过滤进行收集,用水洗涤和在真空(油泵)下干燥,得到75g4-丙烯酰氨基-苯酚。
将4-丙烯酰氨基-苯酚(163mg,1mmol)和单-boc-1,6-己烷二胺(arkpharminc.)(237mg,1.1mmol)溶解在异丙醇(9ml)和水(1ml)的溶液中,并在油浴中在65℃加热48小时。将反应混合物通过旋转蒸发进行浓缩,得到迈克尔加成产物,其不经进一步纯化地用于下一步。
向得自以上步骤的残余物中加入ch2cl2(4ml)和4ml饱和碳酸氢钠水溶液。在室温在搅拌下逐滴加入乙酰氯(0.21ml,3mmol),并将混合物在室温搅拌另外3h。使层分离,并通过旋转蒸发浓缩ch2cl2层。
将残余物溶解在4mlch2cl2中,并在搅拌下逐滴加入2ml4mhcl在二
向以上固体中加入10mlch2cl2和10ml饱和碳酸氢钠水溶液。在搅拌下逐滴加入苯甲酰氯(0.23ml,2mmol),并将混合物在室温搅拌另外3小时。使层分离,并用旋转蒸发器浓缩ch2cl2层。将残余物溶解在2mlmeoh中,并加入2ml5%的naoh在水中的溶液。将混合物在室温搅拌30min(该步骤是除去任何苯甲酰基化的苯酚所必需的)。将混合物用1mhcl中和,并用etoac萃取。将有机层经na2so4干燥,过滤,并通过旋转蒸发除去溶剂。对残余物进行使用5%的meoh在ch2cl2中的溶液的硅胶色谱法,得到170mg纯产物(40%总收率)。
方法2.在搅拌下在氮气氛下向冰冷却的单-boc-1,6-己烷二胺·hcl(20g,79.12mmol)在干燥二氯甲烷(350ml)中的溶液中逐滴加入无水三乙胺(33ml,237.4mmol)。10min以后,在0℃逐滴加入苯甲酰氯(9.64ml,83mmol),并将得到的混合物在室温搅拌过夜。加入水(200ml),并将水层用200ml部分的ch2cl2萃取2次,并将有机萃取物合并,并用水、盐水洗涤,经无水na2so4干燥,过滤并通过旋转蒸发浓缩。将得到的固体用己烷洗涤以除去较少极性的杂质,并将固体不经纯化用于下一步。
将以上固体溶解在100mlch2cl2中,并在0℃在搅拌下逐滴加入300ml20%的tfa在ch2cl2中的溶液,并将得到的混合物在室温搅拌过夜。将反应混合物通过旋转蒸发进行浓缩。将得到的残余物溶解在300ml水中,并用200ml部分的ch2cl2洗涤2次以除去较少极性的杂质。将得到的水层用5%的naoh在水中的溶液中和,并用200ml部分的ch2cl2萃取4次。将有机层合并和经na2so4干燥,过滤并通过旋转蒸发除去溶剂。将得到的粗产物(14.6g,66.4mmol,84%)不经进一步纯化地用于下一步。以此方式得到游离胺,其是下一步(迈克尔加成)所需要的。
将4-丙烯酰氨基-苯酚(8.43g,51.6mmol)和单-苯甲酰基-1,6-己烷二胺(12.5g,56.8mmol)溶解在异丙醇(450ml)和水(50ml)的溶液中,并在65℃油浴中加热48小时。将反应混合物通过旋转蒸发进行浓缩,得到迈克尔加成产物,将其分成2份,并不经进一步纯化用于下一步。
乙酰化.向得自以上步骤的残余物中加入ch2cl2(100ml)、dmf(10ml)和100ml饱和碳酸氢钠水溶液。在室温在搅拌下逐滴加入乙酰氯(3.7ml,52mmol),并将混合物在室温搅拌另外6h。将有机层分离,并将水层用50ml部分的5%的meoh在ch2cl2中的溶液萃取2次。将有机层合并,并通过旋转蒸发浓缩。通过硅胶柱色谱法(1-5%的meoh在ch2cl2中的溶液)纯化残余物,以41%收率得到mps-i糖苷配基(4.5g,10.6mmol)。
戊酰化.向得自以上步骤的残余物中加入ch2cl2(100ml)、dmf(10ml)和100ml饱和碳酸氢钠水溶液。在室温在搅拌下逐滴加入戊酰氯(6.17ml,52mmol),并将混合物在室温搅拌另外6h。将有机层分离,并将水层用50ml部分的5%的meoh在ch2cl2中的溶液萃取2次。将有机层合并,并通过旋转蒸发浓缩。通过硅胶柱色谱法(1-5%的meoh在ch2cl2中的溶液)纯化残余物,以29%收率得到mps-ii糖苷配基(3.5g,7.5mmol)。
mps-i-is-乙酰基-c6
除了使用d5-苯甲酰氯(aldrich)以外,如关于mps-i糖苷配基所述制备内部标准品,mps-i-p-乙酰基-c6。酶产物mps-i-p-乙酰基-c6是mps-i糖苷配基(即,与is相同,但是具有未氘代的苯甲酰基)。
糖苷配基与iduronyl-f的偶联、脱乙酰化和甲酯水解
以下描述了与mps-ii糖苷配基偶联的规程。与mps-i糖苷配基偶联的规程是类似的。
将mps-ii糖苷配基(1.9g,4.06mmol,1当量)、甲基2,3,4-三羟基-iduronosy-1-f(methyl2,3,4-trihydroxy-iduronosy-1-f)(1.23g,3.66mmol,0.9当量)和2,6-二叔丁基-4-甲基吡啶(2.5g,12.2mmol,3当量)在高真空(油泵)下干燥1小时,并溶解于干燥的ch2cl2(80ml,0.05m)中。所有mps-ii糖苷配基在加入bf3-乙醚络合物之前溶解。对于与mps-i糖苷配基的反应,使用更多的ch2cl2来产生0.02m糖苷配基(即使在加入bf3-乙醚络合物以后,并非都溶解)。
在氮气氛下在搅拌下在室温逐滴加入bf3·et2o(5.1ml,40.6mmol,10当量)。已经将反应混合物在室温搅拌2.5h以后,加入150ml饱和nahco3水溶液。将水层用ch2cl2萃取并将有机萃取物合并,并用水、盐水洗涤和经无水na2so4干燥。将溶液过滤并通过旋转蒸发浓缩。通过硅胶柱色谱法(ch2cl2,然后1-4%的meoh在ch2cl2中的溶液)纯化残余物,以65%收率得到产物(1.87g,2.38mmol)。
脱乙酰化.在0℃在氮气氛下在搅拌下向偶联的产物(2.7g,3.44mmol,1当量)在75ml干燥的甲醇(aldrich)中的溶液中逐滴加入0.5m的甲醇钠在甲醇中的溶液(2.75ml,1.38mmol,0.4当量)。将反应混合物在0℃搅拌3h。将反应混合物用ag50w-x8树脂(h+)中和并过滤。通过旋转蒸发浓缩滤液。在硅胶上的柱色谱法(1-6%的meoh在ch2cl2中的溶液)以92%收率得到产物(2.1g,3.19mmol)。
甲酯水解.通过甲酯的脱甲基化制备mps-i-s-乙酰基-c6。对于mps-ii-s-戊酰基-c6,将脱乙酰化的化合物硫酸化,然后将甲酯水解。如关于mps-i-s-乙酰基-c6那样,在没有硫酸化下通过甲酯皂化来制备mps-ii-p-戊酰基-c6。如关于mps-ii-p-戊酰基-c6那样制备mps-ii-is-戊酰基-c6,但是使用含有d5-苯甲酰基的糖苷配基。
在室温将去酰化的化合物(1.5g,2.44mmol,1当量)溶解在150ml水/甲醇(1:1)中。以0.1当量的naoh的增量加入0.1m的氢氧化钠水溶液,直到溶液的ph达到大约8(ph试纸)。通过随着反应进行逐渐添加0.1mnaoh溶液,维持ph(添加约2当量的naoh)。将反应混合物搅拌过夜。将反应混合物用1mhcl中和并通过旋转蒸发浓缩。通过硅胶上的柱色谱法(5%meoh和1%的acoh在ch2cl2中的溶液,然后10%meoh和2%的acoh在ch2cl2中的溶液)纯化残余物,以98%收率得到产物mps-i-s-乙酰基-c6(1.45g,2.41mmol)。
重要的是,从底物除去尽可能多的mps-i酶产物,否则测定空白会更高。可以将底物溶解在ph7的水中,并用etoac萃取,因为产物将较好地萃取。但是,底物(因为它的羧酸酯,是阴离子的)将保留在水中。将1.5gmps-i-s-乙酰基-c6溶解在200ml蒸馏水中,并使用ph计用koh将ph调至接近7。用3个200ml部分的etoac萃取。将水层转移至圆底烧瓶,并放在具有水抽吸和30℃水浴的旋转蒸发器上和旋转蒸发约20min以除去水中的任何etoac。然后,将水层低压冻干以产生终产物,即mps-i-s-乙酰基-c6的钠盐。该规程产生含有mps-i产物的底物。研究了替代性纯化。
在一个替代方案中,将50mgmps-i-s-ac-c6溶解在5ml水中,用稀naoh水溶液调至ph7(ph计),通过涡旋用8ml乙酸乙酯萃取,然后离心以分离各层,重复另外4次(所以共计40ml乙酸乙酯)。将水层低压冻干。mps-i产物的量是非常低的,且是该纯化可接受的。
在另一个替代方案中,使用线性1-10%的meoh在ch2cl2中的溶液梯度历时10min然后达到20%meoh/2%水/ch2cl2,通过快速si柱(10g硅胶)纯化120mgmps-i-s-ac-c6。将材料的峰合并成3批,即峰的第一个三分之一(firstthird)、第二个三分之一(secondthird)和最后一个三分之一(lastthird)。在30℃水浴和水抽吸下通过旋转蒸发除去溶剂。用3个单独批次进行的测定表明,中间一个和最后一个三分之一峰材料含有可接受的量的mps-i-产物。
硫酸化和甲酯水解.将脱乙酰化的化合物(2g,3.04mmol,1当量)溶解在无水meoh(120ml)中并加入二丁基锡(iv)氧化物(1.13g,4.56mmol,1.5当量)。将反应混合物在氮气下在回流下加热1小时,该时间以后,二丁基氧化锡完全溶解。将反应混合物冷却并在真空下浓缩。将残余物与无水甲苯(100ml)一起共蒸发一次以除去痕量的水。将残余物溶解在无水n,n-二甲基甲酰胺(120ml)中。加入三氧化硫-三甲胺复合物(633.8mg,4.56mmol,1.5当量),并将反应混合物在氮气氛下在室温搅拌24h。将反应混合物用meoh(20ml)猝灭。然后将混合物在真空下浓缩。通过硅胶上的柱色谱法(10%meoh和1%的h2o在ch2cl2中的溶液,然后20%meoh和2%的h2o在ch2cl2中的溶液)纯化残余物,以53.6%收率得到硫酸酯化合物(1.2g,1.63mmol)。
在室温将硫酸酯化合物(1g,1.35mmol)溶解在1:1甲醇-水(100ml)中。以0.1当量的naoh的增量加入0.1mnaoh水溶液,直到溶液的ph达到大约8(ph试纸)。通过随着反应进行(每15-30min)逐渐添加0.1mnaoh溶液,维持ph。可能重要的是,不要达到高ph,因为这可能导致硫酸酯的稍微水解。将反应混合物搅拌过夜(加入约2当量naoh),此后将它在真空下浓缩以除去甲醇和水。通过硅胶上的柱色谱法(10%meoh和1%的h2o在ch2cl2中的溶液,然后20%meoh和2%的h2o在ch2cl2中的溶液)纯化残余物,以79%收率得到mps-ii-s(0.77g,1.06mmol)。
通过如下所述的萃取或离子交换色谱法,可以从硫酸化的物质除去未硫酸化的物质。
萃取净化.将得自硅胶的化合物(0.77g,1.06mmol)溶解在纯水(200ml)中,并逐滴加入1mhcl以将ph调至2.7(通过ph计)。用etoac(5×200ml)萃取水层。将剩余的水层转移至圆底烧瓶(其放在具有30℃水浴和水抽吸的旋转蒸发器上)30min以从水层除去痕量的etoac。将水层低压冻干。基于5次etoac萃取的lc-msms表明与第一个萃取物相比在第三个萃取物中未硫酸化的物质的约100倍下降。在第4个和第5个萃取物中的量较低,且与在第3个萃取物中的量类似。
离子交换纯化.使用溶剂a(meoh)和溶剂b(meoh+1m甲酸铵)的akta。以3ml/min使用市售的pharmaciahiload26/10q-sepharose柱。将约10mgmps-ii-is-戊-c6-d5在约0.5ml中注射到柱上并在100%a中保持20min,然后历时30min运行0-100%b的线性梯度,并保持在100%b(在51min洗脱)。使用以上程序(2ml环)注射22mgmps-ii-s-戊-c6在1.5mlmeoh中的溶液。底物在100min洗脱。旋转蒸发出meoh(水泵25-30℃水浴)。将残余物溶解在约10ml水中并加载到预先用约100mlmeoh、然后用约100ml水洗涤的watersc18sep-pak(50g大小)上,用约200ml水洗涤(od280nm接近于0)。将化合物用约200mlmeoh洗脱,额外的meoh具有接近于0的od280,通过旋转蒸发(水泵,30℃水浴)除去甲醇。将残余物溶解在几mlmeoh中并转移至2×5ml玻璃管形瓶(使用更多的meoh完成转移)并进行没有加热的speed-vac过夜。
实施例2
使用mps-i试剂的代表性测定
在该实施例中,描述了使用本发明的mps-i试剂的代表性测定。将这些试剂的结果与其它mps-i试剂进行对比。
下面显示了最初的mps-i反应(blanchard,sophie,sadilek,martin,scott,c.ronald,turecek,frantisek,和gelb,michaelh.(2008)“tandemmassspectrometryforthedirectassayoflysosomalenzymesindriedbloodspots:applicationtoscreeningnewbornsformucopolysaccharidosisi,”clin.chem.,54:2067-2070)。注意到,s、p和is具有boc基团,并且p和is不是化学相同的(p在连接体中具有4个ch2基团,而is具有3个)。
下面显示了替代性的mps-i反应:
注意到不同的糖苷配基具有n-乙酰基,没有boc氨基甲酸酯基。还注意到,内部标准品与产物在化学上相同,但是具有5个在苯甲酰基中的氘。
在如下的并行(side-by-side)酶测定中对比最初的和替代性的mps-i底物:0.5mm底物,在30μl缓冲液中的3.5μm内部标准品(100mm甲酸铵,ph4.0)。加入干燥血斑点的3mm穿孔,并将混合物在37℃在摇动下温育16小时。通过加入120μl乙腈,猝灭反应。将孔离心,并将120μl上清液转移至新孔。通过加入120μl水,稀释样品,并将10μl注射到lc/msms系统上。lc和ms/ms条件如公开的那样(spacil,z.,tatipaka,h.,barcenas,m.,scott,c.r.,turecek,f.,gelb,m.h.(2012)“high-throughputassayof9lysosomalenzymesfornewbornscreening.”clinicalchemistry.,59(3),1530-8561)。还进行空白测定,其中用无血液的滤纸的3mm穿孔替代干燥血斑点。将空白温育并如上处理。
表3.比较性的mps-i测定结果.
1将酶活性表达为每升血液每小时形成的产物的μmol。
2变动系数(cv)是基于6次测定运行,每次运行用得自相同干燥血斑点的不同穿孔进行。
3msms应答是每pmol分析物在串联质谱法通道中测量的离子计数的量。
4血液-无血液测定比率是用干燥血斑点穿孔在测定中测得的酶活性与用无血液穿孔测得的酶活性之比。
从上表可以看出,两种mps-i底物表现出对mps-i酶的类似活性(每升血液每小时生产的μmol产物),但是替代性底物产生的产物在msms检测中的灵敏度高约5倍(每pmol分析物检测到的离子计数)。通过流动注射-msms得到类似的结果。替代性的mps-i产物和内部标准品的msms应答比最初物质高约5倍(未显示)。
将mps-i产物的msms应答与法布里测定产物(下面显示的结构)进行对比。
注意到mps-i产物具有在胺上的乙酰基,而法布里产物具有boc氨基甲酸酯。boc氨基甲酸酯由于异丁烯和co2的损失在msms仪器的电喷射电离源中部分地分解以产生用h替代boc的分解产物。法布里产物产生211离子计数/pmol,而mps-i产物产生500离子计数/pmol。
实施例3
使用mps-ii试剂的代表性测定
在该实施例中,描述了使用本发明的mps-ii试剂的代表性测定。将这些试剂的结果与其它mps-ii试剂进行对比。
下面显示了最初的msp-ii反应(wolfe,b.j.,blanchard,s.,sadilek,m.,scott,c.r.,turecek,f.,gelb,m.h.(2011)“tandemmassspectrometryforthedirectassayoflysosomalenzymesindriedbloodspots:applicationtoscreeningnewbornsformucopolysaccharidosisii(huntersyndrome)”anal.chem.,83:1152-1156)。注意到,s、p和is具有boc基团。
下面显示了替代性的mps-ii反应:
注意到不同的糖苷配基具有n-戊酰基且缺少boc氨基甲酸酯基。还注意到内部标准品与产物在化学上相同,但是具有5个在苯甲酰基中的氘。
在如下的并行酶测定中对比最初的和替代性的mps-ii底物:在30μl缓冲液(100mm甲酸铵,ph4.0,7.5mm乙酸钡(ii),5.0mm乙酸铈(iii))中的1mm底物、5μm内部标准品。加入干燥血斑点的3mm穿孔,并将混合物在37℃在摇动下温育16小时。通过加入200μl44mm柠檬酸,随后加入400μl乙酸乙酯和100μl水,猝灭反应。用吸量管上下混合几次后,将样品离心(10min,在3000rpm)以分离液体层。将200μl部分的上乙酸乙酯层转移至新孔,并通过与无油空气流一起蒸发来除去溶剂。将残余物溶解于100μl甲醇/5mm甲酸铵水溶液(80/20,v/v)中并输入串联质谱仪中。存在钡和铈盐以沉淀存在于干燥血斑点中的游离硫酸盐和磷酸盐,因为这些阴离子会造成mps-ii酶的生产抑制。使用柠檬酸来酸化混合物,使得mps-ii产物和内部标准品的艾杜糖醛酸部分的羧酸酯被质子化并萃取进乙酸乙酯中。还进行了空白测定,其中用无血液的滤纸的3mm穿孔替代干燥血斑点。将空白温育并如上处理。
表4.可比较的mps-ii测定结果.
1将酶活性表达为每升血液每小时形成的产物的μmol。
2变动系数(cv)是基于6次测定运行,每次运行用得自相同干燥血斑点的不同穿孔进行。
3msms应答是每pmol分析物在串联质谱法通道中测量的离子计数的量。
4血液-无血液测定比率是用干燥血斑点穿孔在测定中测得的酶活性与用无血液穿孔测得的酶活性之比。
从上表可以看出,两种mps-ii底物表现出对mps-ii酶的类似活性(每升血液每小时生产的μmol产物),但是替代性底物产生的产物在msms检测中的灵敏度高约10倍(每pmol分析物检测到的离子计数)。
实施例4
代表性mps-iva底物和酶产物的合成
在该实施例中,描述了代表性mps-iva底物试剂的合成。在图2中显示了mps-iva底物的合成的一般方案。
二乙酸-(2r,3r,4r,5r,6s)-5-乙酰氨基-2-(乙酰氧基甲基)-6-(4-硝基苯氧基)四氢-2h-吡喃-3,4-二酯(1)
将吡啶(60ml)加入氮气逆向清洗的含有d-半乳糖胺盐酸盐(5g,23.2mmol)的烧瓶中,并将得到的浆在冰浴上冷却。向冷却的混合物中逐滴加入乙酸酐(25g,245mmol),并将其温热至室温,随后在该温度搅拌16小时。将反应混合物通过加入甲醇(15ml)进行猝灭,并搅拌20分钟。将得到的混合物在减压下浓缩,并借助于温热所述混合物将残余物溶解在20%的甲醇在氯仿中的溶液中。将该溶液用1nhcl溶液、随后用盐水溶液洗涤。将得到的有机层使用无水硫酸钠干燥并在减压下浓缩。将残余物溶解在氮气逆向清洗的配有滴液漏斗的烧瓶中。将无水二氯甲烷(100ml)加入该残余物中,并将得到的浆在冰浴上冷却。在滴液漏斗中,将氯化钛(6.5g,42.1mmol)溶解在无水二氯甲烷(40ml)中,并将得到的溶液逐滴加入冷却的溶液中。将反应混合物在油浴中温热至50℃并在该温度放置搅拌48小时。将反应混合物在冰浴上冷却回去,并在剧烈摇动下逐滴加入饱和的碳酸氢钠溶液。将得到的混合物在二氯甲烷和饱和的碳酸氢钠溶液之间萃取。将有机层用无水硫酸钠干燥并在减压下浓缩。将得到的残余物溶解在丙酮(60ml)中,并缓慢地加入到4-硝基苯酚(16.1g,116mmol)在丙酮(130ml)和4nkoh水溶液(23.2ml)中的溶液中。将反应物在室温搅拌48小时并在减压下浓缩至小于20ml。将该溶液在1nnaoh和氯仿之间萃取。将有机层用无水硫酸钠干燥并在减压下浓缩。使用3%的甲醇在二氯甲烷中的溶液作为洗脱混合物,通过硅胶快速色谱法纯化如此得到的粗产物。将通过tlc确定含有期望的化合物的级分合并,并在减压下浓缩以得到1(3.29g,30%)。1hnmr(300mhz,cdcl3)δ8.20(d,j=9.1hz,2h),7.09(d,j=9.1hz,2h),5.61(d,j=8.0hz,1h),5.56-5.39(m,3h),4.32-4.07(m,4h),2.18(s,3h),2.07(s,3h),2.04(s,3h),1.97(s,3h).[m+na]+的ms(esi+);计算:491.1,实测:491.2。
n-(5-(n-(3-((4-(((2s,3r,4r,5r,6r)-3-乙酰氨基-4,5-二羟基-6-(羟基甲基)四氢-2h-吡喃-2-基)氧基)苯基)氨基)-3-氧代丙基)戊烷酰氨基)戊基)苯甲酰胺(2)
向在冰浴上冷却的1(3.5g,7.47mmol)在无水甲醇(90ml)中的溶液中,逐滴加入0.5m甲醇钠在甲醇(3ml,1.50mmol)中的溶液,并将其温热至室温。2小时以后,将甲酸(0.1ml)加入反应混合物中并在减压下浓缩至干燥。向得到的残余物中加入甲醇(135ml)、水(15ml)和10%活性炭载钯(125mg),并在氢气氛下在室温搅拌16小时。将水逐滴加入反应混合物中,直到整个白色残余物完全溶解。将反应混合物过滤,并将滤液在冰浴上冷却。向它加入吡啶(2ml),随后逐滴加入丙烯酰氯(2.1g,23.2mmol)在二氯甲烷(50ml)中的溶液。将反应物在冰浴上搅拌30分钟,然后温热至室温并继续2小时。将碳酸钠粉末(3.0g)加入反应混合物中并搅拌15分钟和过滤。将滤液在减压下浓缩并在高真空下进一步干燥。将残余物溶解在2-丙醇(50ml)和水(6.6ml)混合物中,并向它加入n-(5-氨基戊基)苯甲酰胺(2.0g,9.69mmol),并在65℃搅拌40小时。将反应混合物冷却至室温,并向它加入甲醇(25ml)。在冰浴上冷却该混合物以后,加入三乙胺(2.5ml),随后逐滴加入戊酰氯(2.7g,22.4mmol)在二氯甲烷(50ml)中的溶液。将反应物在冰浴上搅拌30分钟,然后温热至室温并继续16小时。将反应混合物在减压下浓缩,并使用15%的甲醇在二氯甲烷中的溶液作为洗脱混合物通过硅胶快速色谱法进行纯化,得到2(2.96g,60%)。1hnmr(300mhz,meod)δ7.84(d,j=7.1hz,2h),7.59-7.35(m,5h),7.09-6.91(m,2h),5.00(d,j=8.4hz,1h),4.28-4.09(m,1h),3.97-3.55(m,7h),3.46-3.24(m,4h),2.72-2.51(m,2h),2.49-2.28(d,j=7.3hz,2h),2.02(s,3h),1.78-1.49(m,6h),1.49-1.22(m,4h),0.94(t,j=7.1hz,3h).[m+h]+的ms(esi+);计算:657.3,实测:657.5。
((2r,3r,4r,5r,6s)-5-乙酰氨基-6-(4-(3-(n-(5-苯甲酰氨基戊基)-戊烷酰氨基)丙烷酰氨基)苯氧基)-3,4-二羟基四氢-2h-吡喃-2-基)甲基硫酸钠(3)
在氮气下向化合物2(32mg,0.048mmol)中加入无水吡啶(1ml)。向该溶液中加入三氧化硫-吡啶复合物(18mg,0.113mmol)并在室温搅拌5小时。将反应物通过加入甲醇(0.3ml)进行猝灭并搅拌30分钟。将反应混合物在减压下浓缩并再溶解在水中,并使用水-甲醇梯度系统进行反相(c18)hplc纯化以得到3(23mg,63%)。1hnmr(300mhz,meod)δ7.81(d,j=7.3hz,2h),7.57-7.39(m,5h),7.00(dd,j=9.0,2.4hz,2h),4.93(d,j=8.5hz,1h),4.38-4.12(m,3h),3.95(t,j=4.5hz,2h),3.83-3.61(m,3h),3.49-3.34(m,4h),2.72-2.54(m,2h),2.50-2.30(m,2h),1.98(s,3h),1.78-1.49(m,6h),1.49-1.24(m,4h),1.02-0.84(m,3h).[m-na+]-的ms(esi-);计算:735.3,实测:735.5。
n-(6-(n-(3-((4-(((2s,3r,4r,5r,6r)-3-乙酰氨基-4,5-二羟基-6-(羟基甲基)四氢-2h-吡喃-2-基)氧基)苯基)氨基)-3-氧代丙基)己烷酰氨基)己基)苯甲酰胺(4)
向在冰浴上冷却的1(0.27g,0.576mmol)在无水甲醇(9ml)中的溶液中,逐滴加入0.5m甲醇钠在甲醇中的溶液(0.3ml,0.15mmol),并将其温热至室温。2小时以后,将甲酸(10μl)加入反应混合物中并在减压下浓缩至干燥。向得到的残余物中加入甲醇(13.5ml)、水(1.5ml)和10%活性炭载钯(12.5mg),并在氢气氛下在室温搅拌16小时。将水逐滴加入反应混合物中,直到整个白色残余物完全溶解。将反应混合物过滤,并将滤液在冰浴上冷却。向它加入吡啶(0.16ml),随后逐滴加入丙烯酰氯(0.16g,1.76mmol)在二氯甲烷(4ml)中的溶液。将反应物在冰浴上搅拌30分钟,然后温热至室温并继续2小时。将碳酸钠粉末(0.3g)加入反应混合物中并搅拌15分钟和过滤。将滤液在减压下浓缩并在高真空下进一步干燥。将残余物溶解在2-丙醇(6.3ml)和水(0.7ml)混合物中,并向它加入n-(6-氨基己基)苯甲酰胺(0.17g,0.77mmol),并在65℃搅拌40小时。将反应混合物冷却至室温,并向它加入甲醇(8ml)。在冰浴上冷却该混合物以后,加入三乙胺(0.25ml),随后逐滴加入己酰氯(0.24g,1.78mmol)在二氯甲烷(4ml)中的溶液。将反应物在冰浴上搅拌30分钟,然后温热至室温并继续16小时。将反应混合物在减压下浓缩,并使用15%的甲醇在二氯甲烷中的溶液作为洗脱混合物通过硅胶快速色谱法进行纯化,得到4(0.23g,58%)。1hnmr(300mhz,meod)δ7.80(d,j=6.9hz,2h),7.56-7.39(m,5h),6.99(d,j=9.1hz,2h),4.96(d,j=8.4hz,1h),4.17(dd,j=10.7,8.4hz,1h),3.96-3.51(m,7h),3.44-3.33(m,4h),2.69-2.50(m,2h),2.48-2.28(m,2h),1.98(s,3h),1.72-1.49(m,6h),1.49-1.21(m,9h),0.89(dt,j=8.7,4.8hz,3h)。[m+h]+的ms(esi+);计算:685.4,实测:685.5。
((2r,3r,4r,5r,6s)-5-乙酰氨基-6-(4-(3-(n-(6-苯甲酰氨基己基)-己烷酰氨基)丙烷酰氨基)苯氧基)-3,4-二羟基四氢-2h-吡喃-2-基)甲基硫酸钠(5)
在氮气下向化合物4(99mg,0.144mmol)中加入无水吡啶(5ml)。向该溶液中加入三氧化硫-吡啶复合物(34mg,0.214mmol),并在室温搅拌5小时。将反应物通过加入甲醇(0.5ml)进行猝灭并搅拌30分钟。将反应混合物在减压下浓缩并再溶解在水中,并使用水-甲醇梯度系统进行反相(c18)hplc纯化以得到3(36mg,32%)。1hnmr(300mhz,meod)δ7.81(d,j=6.9hz,2h),7.59-7.35(m,5h),7.00(d,j=9.0hz,2h),4.93(d,j=8.4hz,1h),4.32-4.10(m,3h),4.03-3.90(m,2h),3.83-3.60(m,3h),3.44-3.33(m,4h),2.61(q,j=7.0hz,2h),2.49-2.29(m,2h),1.98(s,3h),1.72-1.49(m,6h),1.48-1.22(m,9h),0.98-0.81(m,3h).[m-na+]-的ms(esi-);计算:763.3,实测:763.7。
实施例5
代表性mps-vi底物和酶产物的合成
在该实施例中,描述了代表性mps-vi底物和酶产物试剂的合成。在图3中显示了mps-vi底物的合成的一般方案。
mps-vi底物(己烷酰氨基).在下面描述了并在图4中图解了代表性mps-vi底物(己烷酰氨基)的制备。
n-(5-(n-(3-((4-羟基苯基)氨基)-3-氧代丙基)己烷酰氨基)戊基)-苯甲酰胺(3)
在恒定搅拌下将n-(5-氨基戊基)苯甲酰胺(180mg,0.872mmol)和n-(4-羟基苯基)丙烯酰胺(171mg,1.05mmol)在异丙醇(7.8ml)和水(0.87ml)中的溶液加热至65℃保持24小时。将反应混合物冷却至室温,并在减压下和进一步在高真空下浓缩。向该粗制浓缩物中加入无水n,n-二甲基甲酰胺(dmf)(3.0ml)和三乙胺(220mg,2.17mmol)并彻底溶解。将该溶液冷却至0℃,并逐滴加入己酰氯(234mg,1.74mmol)和温热至室温和搅拌2小时。通过加入饱和碳酸氢钠溶液猝灭反应,并用dcm/甲醇(4:1)萃取反应混合物。将有机层用水进一步洗涤和用无水硫酸钠干燥。将有机层在减压下浓缩至干燥,并加入甲醇(3.0ml)和再溶解。向该溶液中逐滴加入5%的氢氧化钠水溶液(3.0ml)并在室温搅拌2小时。将反应物用1nhcl溶液酸化(如ph试纸指示的),并用dcm/甲醇(4:1)萃取。将有机层在减压下浓缩,并对残余物进行硅胶柱色谱法和用5%的甲醇在dcm中的溶液洗脱,产生3(151mg,37%)。1hnmr(300mhz,meod)δ8.47(s,1h),7.81(d,j=7.7hz,2h),7.57-7.40(m,3h),7.36-7.27(m,2h),6.78-6.67(m,2h),3.69(dt,j=18.7,6.9hz,2h),3.46-3.33(m,4h),2.60(q,j=7.0hz,2h),2.47-2.29(m,2h),1.63(ddd,j=11.6,10.6,5.7hz,6h),1.48-1.22(m,6h),0.89(td,j=6.6,2.6hz,3h)。[m+na]+的ms(esi+);计算:490.3,实测:490.6。
n-(5-(n-(3-((4-(((2s,3r,4r,5r,6r)-3-乙酰氨基-4,5-二羟基-6-(羟基甲基)四氢-2h-吡喃-2-基)氧基)苯基)氨基)-3-氧代丙基)己烷酰氨基)戊基)苯甲酰胺(4)
向3(73mg,0.156mmol)和二乙酸-(2r,3r,4r,5r)-5-乙酰氨基-2-(乙酰氧基甲基)-6-氯四氢-2h-吡喃-3,4-二酯(114mg,0.312mmol)在无水dmf(0.7ml)中的溶液中加入碳酸铯(152mg,0.466mmol),并在室温搅拌6小时。然后将反应混合物在水和dcm之间萃取,并将有机层进一步用水洗涤,用无水硫酸钠干燥并在减压下浓缩。使用4%的甲醇在dcm中的溶液作为洗脱液,通过快速硅胶柱色谱法纯化得到的粗制物,得到全乙酰化的中间体。nmr光谱法指示全乙酰化的中间体已经与起始原料3一起共洗脱。该混合物不经进一步纯化用于下一个脱乙酰化步骤。在0℃向以上混合物在无水甲醇中的溶液(5.0ml)中逐滴加入0.5m的甲醇钠在甲醇中的溶液(200μl),并在室温搅拌2小时。通过加入甲酸(100μl)猝灭反应,并进行半-制备型反相hplc纯化(梯度水/甲醇系统),得到4(23mg,22%)。1hnmr(300mhz,meod)δ7.85-7.77(m,2h),7.58-7.38(m,5h),6.99(dd,j=9.1,2.6hz,2h),4.96(dd,j=8.4,1.3hz,1h),4.17(dd,j=10.7,8.4hz,1h),3.90(d,j=3.1hz,1h),3.84-3.60(m,6h),3.38(dd,j=11.1,6.8hz,4h),2.61(q,j=7.1hz,2h),2.47-2.29(m,2h),1.98(s,3h),1.74-1.50(m,6h),1.47-1.21(m,6h),0.89(td,j=6.6,3.3hz,3h)。[m+na]+的ms(esi+);计算:693.3,实测:693.4。
(2r,3r,4r,5r,6s)-5-乙酰氨基-6-(4-(3-(n-(5-苯甲酰氨基戊基)-己烷酰氨基)丙烷酰氨基)苯氧基)-4-羟基-2-(羟基甲基)四氢-2h-吡喃-3-基硫酸钠(5)
向冷却(0℃)的4(22mg,32.8μmol)在无水吡啶(0.5ml)中的溶液中,加入苯甲酰氯(7.7μl,65.6μmol)。在室温1小时以后,将溶液冷却回0℃,并加入另一部分的苯甲酰氯(7.7μl,65.6μmol),在室温搅拌2小时。通过加入甲醇(200μl)来猝灭反应,并搅拌另外30分钟。将得到的混合物在减压下浓缩,并使用4%的甲醇在dcm中的溶液作为洗脱液通过快速硅胶柱色谱法纯化。将期望的级分在减压下和进一步在高真空下浓缩。将得到的残余物溶解在无水吡啶(1.0ml)中,并在室温向它加入三氧化硫-吡啶复合物(17mg,109μmol)。将得到的混合物加热至45℃保持3小时,随后加入甲醇(0.5ml)和搅拌另外10分钟。将反应混合物在减压下和进一步在高真空下浓缩。将得到的残余物再溶解在无水甲醇(6.0ml)中并冷却至0℃。向该冷却的溶液中逐滴加入0.5m的甲醇钠在甲醇中的溶液(0.8ml),并搅拌16小时。通过加入1m磷酸二氢钠水溶液(1.0ml)来猝灭反应,并进行半-制备型反相hplc纯化(梯度水/甲醇系统)以产生5(12mg,47%)。1hnmr(300mhz,meod)δ7.85-7.76(m,2h),7.58-7.38(m,5h),7.05-6.92(m,2h),5.00(dd,j=8.4,1.2hz,1h),4.75(d,j=3.1hz,1h),4.15(dd,j=10.9,8.4hz,1h),3.95-3.60(m,6h),3.38(dt,j=11.2,5.6hz,4h),2.62(dd,j=15.9,6.9hz,2h),2.47-2.29(m,2h),1.97(s,3h),1.76-1.51(m,6h),1.47-1.21(m,6h),0.89(td,j=6.6,3.4hz,3h)。[m-na+]-的ms(esi-);计算:749.3,实测:749.5。
mps-vi底物(戊烷酰氨基).在下面描述了并在图4中图示了代表性mps-vi底物(戊烷酰氨基)的制备。
n-(5-(n-(3-((4-羟基苯基)氨基)-3-氧代丙基)戊烷酰氨基)戊基)-苯甲酰胺.将n-(5-氨基戊基)苯甲酰胺(207mg,1.00mmol)和n-(4-羟基苯基)丙烯酰胺(197mg,1.21mmol)在异丙醇(9.0ml)和水(1.0ml)中的溶液在恒定搅拌下加热至65℃保持24小时。将反应混合物冷却至室温,并在减压下和进一步在高真空下浓缩。向该粗制浓缩物中加入无水n,n-二甲基甲酰胺(dmf)(3.0ml)和三乙胺(253mg,2.50mmol),并彻底溶解。将该溶液冷却至0℃并逐滴加入戊酰氯(241mg,2.00mmol),并温热至室温和搅拌2小时。通过加入饱和碳酸氢钠溶液来猝灭反应,并用dcm/甲醇(4:1)萃取反应混合物。将有机层进一步用水洗涤和用无水硫酸钠干燥。将有机层在减压下浓缩至干燥并加入甲醇(3.0ml)和再溶解。向该溶液中逐滴加入5%的氢氧化钠水溶液(3.0ml)并在室温搅拌2小时。将反应物用1nhcl溶液酸化(如ph试纸指示的),并用dcm/甲醇(4:1)萃取。将有机层在减压下浓缩,并对残余物进行硅胶柱色谱法用5%的甲醇在dcm中的溶液洗脱,以产生标题化合物(203mg,45%)。1hnmr(300mhz,meod)δ8.45(s,1h),7.83-7.78(m,2h),7.56-7.40(m,3h),7.34-7.26(m,2h),6.76-6.68(m,2h),3.69(dt,j=19.4,6.9hz,2h),3.44-3.37(m,4h),2.60(dd,j=14.7,7.4hz,2h),2.47-2.30(m,2h),1.75-1.49(m,6h),1.47-1.26(m,4h),0.91(t,j=7.3hz,3h)。[m+na]+的ms(esi+);计算:476.3,实测:476.5。
n-(5-(n-(3-((4-(((2s,3r,4r,5r,6r)-3-乙酰氨基-4,5-二羟基-6-(羟基甲基)-四氢-2h-吡喃-2-基)氧基)苯基)氨基)-3-氧代丙基)戊烷酰氨基)戊基)苯甲酰胺.向n-(5-(n-(3-((4-羟基苯基)氨基)-3-氧代丙基)戊烷酰氨基)戊基)-苯甲酰胺(148mg,0.326mmol)和二乙酸-(2r,3r,4r,5r)-5-乙酰氨基-2-(乙酰氧基甲基)-6-氯四氢-2h-吡喃-3,4-二酯(239mg,0.653mmol)在dcm(0.4ml)中的溶液中加入四丁基硫酸氢铵(110mg,0.324mmol)和2m氢氧化钠水溶液(0.4ml),并在室温搅拌3小时。向该反应混合物中加入另一部分的二乙酸酯(90mg,0.246mmol),并搅拌另外13小时。然后将反应混合物在水和dcm之间萃取,并将有机层进一步用水洗涤,用无水硫酸钠干燥并在减压下浓缩。使用4%的甲醇在dcm中的溶液作为洗脱液,通过快速硅胶柱色谱法纯化得到的粗制物,得到全乙酰化的中间体。nmr光谱法指示全乙酰化的中间体已经与起始原料一起共洗脱。该混合物不经进一步纯化用于下一个脱乙酰化步骤。在0℃向以上混合物在无水甲醇(5.0ml)中的溶液中逐滴加入0.5m的甲醇钠在甲醇中的溶液(200μl),并在室温搅拌2小时。通过加入甲酸(100μl)猝灭反应,并进行半-制备型反相hplc纯化(梯度水/甲醇系统),得到标题化合物(29mg,14%)。1hnmr(300mhz,meod)δ7.83(d,j=7.1hz,2h),7.59-7.35(m,5h),7.06-6.90(m,2h),4.99(d,j=8.4hz,1h),4.27-4.11(m,1h),3.96-3.55(m,7h),3.46-3.35(m,4h),2.71-2.51(m,2h),2.49-2.27(m,2h),2.01(s,3h),1.76-1.49(m,6h),1.37(dd,j=14.6,7.2hz,4h),0.93(t,j=7.2hz,3h)。[m+na]+的ms(esi+);计算:679.3,实测:679.7。
(2r,3r,4r,5r,6s)-5-乙酰氨基-6-(4-(3-(n-(5-苯甲酰氨基戊基)-戊烷酰氨基)丙烷酰氨基)苯氧基)-4-羟基-2-(羟基甲基)四氢-2h-吡喃-3-基硫酸钠.向冷却(0℃)的n-(5-(n-(3-((4-(((2s,3r,4r,5r,6r)-3-乙酰氨基-4,5-二羟基-6-(羟基甲基)-四氢-2h-吡喃-2-基)氧基)苯基)氨基)-3-氧代丙基)戊烷酰氨基)戊基)苯甲酰胺(25mg,38.1μmol)在无水吡啶(0.5ml)中的溶液中,加入苯甲酰氯(4.9μl,41.9μmol)。在室温1小时以后,将溶液冷却回0℃,并加入另一部分的苯甲酰氯(9.4μl,80.4μmol),并在室温搅拌2小时。将反应物在1mhcl溶液和氯仿之间萃取。将氯仿层进一步用水和盐水溶液(1:1)的混合物洗涤。将有机层浓缩,并使用5%的甲醇在dcm中的溶液作为洗脱液通过快速硅胶柱色谱法纯化。将期望的级分在减压下并进一步在高真空下浓缩。将得到的残余物溶解在无水吡啶中,并在室温向它加入三氧化硫-吡啶复合物(8.3mg,52.1μmol)。将得到的混合物加热至45℃保持3小时,随后加入甲醇(0.5ml)并搅拌另外10分钟。将反应混合物在减压下和进一步在高真空下浓缩。将得到的残余物再溶解在无水甲醇(5.0ml)中并冷却至0℃。向该冷却的溶液中逐滴加入0.5m的甲醇钠在甲醇中的溶液(0.5ml),并搅拌16小时。将反应通过加入1m磷酸二氢钠水溶液(1.0ml)进行猝灭,并进行半-制备型反相hplc纯化(梯度水/甲醇系统)以产生标题化合物(5.8mg,20%)。1hnmr(300mhz,meod)δ7.80(dd,j=7.0,1.2hz,2h),7.58-7.38(m,5h),7.03-6.94(m,2h),5.01(dd,j=8.4,1.1hz,1h),4.75(d,j=3.1hz,1h),4.13(dd,j=10.9,8.4hz,1h),3.95-3.61(m,6h),3.45-3.34(m,4h),2.61(dd,j=16.1,7.0hz,2h),2.47-2.31(m,2h),1.97(s,3h),1.73-1.49(m,6h),1.43-1.23(m,5h),0.91(td,j=7.3,2.2hz,3h)。[m-na+]-的ms(esi-);计算:735.3,实测:735.4。
mps-vi产物.在下面描述了并在图5中图示了代表性mps-vi产物的制备。
二乙酸-(2r,3r,4r,5r,6s)-5-乙酰氨基-2-(乙酰氧基甲基)-6-(4-硝基苯氧基)四氢-2h-吡喃-3,4-二酯(1)
将吡啶(60ml)加入氮气逆向清洗的含有d-半乳糖胺盐酸盐(5g,23.2mmol)的烧瓶中,并将得到的浆在冰浴上冷却。向冷却的混合物中逐滴加入乙酸酐(25g,245mmol),并将其温热至室温,随后在该温度搅拌16小时。将反应混合物通过加入甲醇(15ml)进行猝灭,并搅拌20分钟。将得到的混合物在减压下浓缩,并通过温热所述混合物将残余物溶解在20%的甲醇在氯仿中的溶液中。将该溶液用1nhcl溶液、随后用盐水溶液洗涤。将得到的有机层使用无水硫酸钠干燥并在减压下浓缩。将残余物溶解在氮气逆向清洗的配有滴液漏斗的烧瓶中。将无水二氯甲烷(100ml)加入该残余物中,并将得到的浆在冰浴上冷却。在滴液漏斗中,将氯化钛(6.5g,42.1mmol)溶解在无水二氯甲烷(40ml)中,并将得到的溶液逐滴加入冷却的溶液中。将反应混合物在油浴中温热至50℃,并在该温度搅拌48小时。将反应混合物在冰浴上冷却回去,并在剧烈摇动下逐滴加入饱和碳酸氢钠溶液。将得到的混合物在二氯甲烷和饱和碳酸氢钠溶液之间萃取。将有机层用无水硫酸钠干燥并在减压下浓缩。将得到的残余物溶解在丙酮(60ml)中,并缓慢地加入4-硝基苯酚(16.1g,116mmol)在丙酮(130ml)和4nkoh水溶液(23.2ml)中的溶液中。将反应物在室温搅拌48小时并在减压下浓缩至小于20ml。将该溶液在1nnaoh和氯仿之间萃取。将有机层用无水硫酸钠干燥并在减压下浓缩。使用3%的甲醇在二氯甲烷中的溶液作为洗脱混合物,通过硅胶快速色谱法纯化如此得到的粗产物。将含有期望的化合物的级分(通过tlc确定)合并,并在减压下浓缩,得到1(3.29g,30%)。1hnmr(300mhz,cdcl3)δ8.20(d,j=9.1hz,2h),7.09(d,j=9.1hz,2h),5.61(d,j=8.0hz,1h),5.56-5.39(m,3h),4.32-4.07(m,4h),2.18(s,3h),2.07(s,3h),2.04(s,3h),1.97(s,3h).[m+na]+的ms(esi+);计算:491.1,实测:491.2。
n-(5-(n-(3-((4-(((2s,3r,4r,5r,6r)-3-乙酰氨基-4,5-二羟基-6-(羟基甲基)四氢-2h-吡喃-2-基)氧基)苯基)氨基)-3-氧代丙基)戊烷酰氨基)戊基)苯甲酰胺(2)
向在冰浴上冷却的1(3.5g,7.47mmol)在无水甲醇(90ml)中的溶液中,逐滴加入0.5m甲醇钠在甲醇中的溶液(3ml,1.50mmol),并将其温热至室温。2小时以后,将甲酸(0.1ml)加入反应混合物中并在减压下浓缩至干燥。向得到的残余物中加入甲醇(135ml)、水(15ml)和10%活性炭载钯(125mg),并在氢气氛下在室温搅拌16小时。将水逐滴加入反应混合物中,直到整个白色残余物完全溶解。将反应混合物过滤,并将滤液在冰浴上冷却。向它加入吡啶(2ml),随后逐滴加入丙烯酰氯(2.1g,23.2mmol)在二氯甲烷(50ml)中的溶液。将反应物在冰浴上搅拌30分钟,然后温热至室温并继续2小时。将碳酸钠粉末(3.0g)加入反应混合物中,并搅拌15分钟和过滤。将滤液在减压下浓缩并在高真空下进一步干燥。将残余物溶解在2-丙醇(50ml)和水(6.6ml)混合物中,并向它加入n-(5-氨基戊基)苯甲酰胺(2.0g,9.69mmol),并在65℃搅拌40小时。将反应混合物冷却至室温,并向它加入甲醇(25ml)。在冰浴上冷却该混合物以后,加入三乙胺(2.5ml),随后逐滴加入戊酰氯(2.7g,22.4mmol)在二氯甲烷(50ml)中的溶液。将反应物在冰浴上搅拌30分钟,然后温热至室温并继续16小时。将反应混合物在减压下浓缩,并使用15%的甲醇在二氯甲烷中的溶液作为洗脱混合物进行硅胶快速色谱法纯化,得到2(2.96g,60%)。1hnmr(300mhz,meod)δ7.84(d,j=7.1hz,2h),7.59-7.35(m,5h),7.09-6.91(m,2h),5.00(d,j=8.4hz,1h),4.28-4.09(m,1h),3.97-3.55(m,7h),3.46-3.24(m,4h),2.72-2.51(m,2h),2.49-2.28(d,j=7.3hz,2h),2.02(s,3h),1.78-1.49(m,6h),1.49-1.22(m,4h),0.94(t,j=7.1hz,3h)。[m+h]+的ms(esi+);计算:657.3,实测:657.5。
实施例6
使用mps-vi试剂的代表性测定
在该实施例中,描述了使用本发明的mps-vi试剂的代表性测定。将这些试剂的结果与其它mps-vi试剂进行对比。
下面显示了最初的msp-vi反应(duffey,t.a.,sadilek,m.,scott,c.r.,turecek,f.,gelb,m.h.(2010)“tandemmassspectrometryforthedirectassayoflysosomalenzymesindriedbloodspots:applicationtoscreeningnewbornsformucopolysaccharidosisvi(maroteaux-lamysyndrome)”,anal.chem.,82:9587-9591)。注意到,s、p和is具有boc基团,并且p和is不是在化学上相同的(p在连接体中具有6个ch2基团,而is具有5个)。
下面显示了替代性的mps-vi反应:
注意到不同的糖苷配基具有n-戊酰基基团,不具有boc氨基甲酸酯基。还注意到内部标准品与产物在化学上相同,但是具有5个在苯甲酰基中的氘。
在如下的并行酶测定中对比了最初的和替代性的mps-vi底物:在30μl缓冲液(100mm甲酸铵,ph4.0,7.5mm乙酸钡(ii),5.0mm乙酸铈(iii))中的1mm底物、10μm内部标准品。加入干燥血斑点的3mm穿孔,并将混合物在37℃在摇动下温育16小时。通过加入deae纤维素(whatmannde52)的水性悬浮液(1:1)(100μl),随后加入400μl乙酸乙酯,猝灭反应。将混合物通过上下抽吸几次进行混合,然后离心(10min,在3000rpm)以分离液体层和沉淀de52。将300μl部分的上乙酸乙酯层转移至新孔,并通过与无油空气流一起蒸发来除去溶剂。将残余物溶解于100μl甲醇/5mm甲酸铵水溶液(80/20,v/v)中并输入串联质谱仪中。存在钡和铈盐以沉淀存在于干燥血斑点中的游离硫酸盐和磷酸盐,因为这些阴离子会造成mps-vi酶的生产抑制。加入de52来捕集剩余的底物,使得仅产物和内部标准品(其为电荷中性的)被萃取进乙酸乙酯中。还进行空白测定,其中用无血液的滤纸的3mm穿孔替换干燥血斑点。将空白温育并如上处理。
表5.可比较的mps-vi测定结果.
1将酶活性表达为每升血液每小时形成的产物的μmol。
2变动系数(cv)是基于6次测定运行,每次运行用得自相同干燥血斑点的不同穿孔进行。
3msms应答是每pmol分析物在串联质谱法通道中测量的离子计数的量。
4血液-无血液测定比率是用干燥血斑点穿孔在测定中测得的酶活性与用无血液穿孔测得的酶活性之比。
从上表可以看出,两种mps-vi底物表现出对mps-vi酶的类似活性(每升血液每小时生产的μmol产物),但是替代性底物产生的产物在msms检测中的灵敏度高约10倍(每pmol分析物检测到的离子计数)。血液-无血液测定应答的改善可能是由于在替代性mps-vi底物中作为污染物的产物的较低量,因为新底物更易于以无产物形式产生。
实施例7
关于mps-ii的代表性硫酸酯酶测定
在该实施例中,描述了本发明的代表性测定(关于艾杜糖醛酸2-硫酸酯酶的测定,该酶在亨特综合征(huntersyndrome)(mps-ii)中缺乏)。
该反应中的第一步如上所述(也参见wo2009/026252(pct/us2008/073516)、wo2010/081163(pct/us2010/020801)、wo2012/027612(pct/us2011/049224)和wo2013/070953(pct/us2012/064205))。
在测定中,将第二种酶α-l-艾杜糖醛酸酶(糖原水解酶)加入测定混合液中,其通过除去艾杜糖醛酸残基剩下糖苷配基而将最初的mps-ii产物转化成最终的mps-ii产物。艾杜糖醛酸酶可以存在于与干燥血斑点一起温育的测定混合液中,或它可以在与干燥血斑点第一次温育、随后的第二个温育时段以后加入。加入的艾杜糖醛酸酶的量足以将最初的mps-ii产物转化成最终的mps-ii产物。测定混合液还含有最初的mps-ii内部标准品(与最初的mps-ii产物相同,例如,具有5个在苯甲酰基中的氘),其被艾杜糖醛酸酶转化成最终的mps-ii内部标准品。通过串联质谱法检测最终的mps-ii产物和最终的mps-ii内部标准品,所述串联质谱法使得可以定量最终的mps-ii产物的量。通常,用有机溶剂萃取反应混合物以造成最终的mps-ii内部标准品和最终的mps-ii产物以相对无盐形式分配进有机溶剂相中。通过蒸发除去有机溶剂,将残余物溶解在溶剂中,并将溶剂注射进串联质谱仪中。通过多重反应监测来检测分析物。
使用不包括第二种酶(即,糖原水解酶)的测定和使用得自新生儿筛查卡的干燥血斑点的3mm穿孔,在12-18小时的温育时间以后通常观察到最初的mps-ii产物的10,000-30,000离子计数。使用本发明的用艾杜糖醛酸酶的测定,通常观察到最终的mps-ii产物的100万-500万离子计数。因而,测定灵敏度已经提高了约100倍。本发明的方法的第二个优点是,在以前的测定中,进入质谱仪电喷射电离源的剩余mps-ii底物由于在所述源中的加热而经历某种程度的去硫酸化。这会通过产生产物信号而增加测定背景,所述产物信号独立于艾杜糖醛酸2-硫酸酯酶的作用。对于本发明的测定,该去硫酸化无足轻重,因为要检测的产物是糖苷配基(最终的mps-ii产物)。
使用的艾杜糖醛酸酶是通过在哺乳动物细胞中过表达得到的人酶。可以使用任何艾杜糖醛酸酶,只要它不会作用于mps-ii底物(即,仅在已经从艾杜糖醛酸除去硫酸酯以后切割糖苷键)。
实施例8
关于mps-vi的代表性硫酸酯酶测定
用在mps-vi的代表性测定中的合适的第二种酶是细菌n-乙酰基半乳糖胺酶,且被用于在mps-vi酶从糖的4-位除去硫酸酯以后从它的糖苷配基释放n-乙酰基-半乳糖胺。这会使测定灵敏度提高约20倍。其它合适的酶包括细菌n-乙酰基己糖胺酶。
实施例9
关于mps-iva的代表性硫酸酯酶测定
用在mps-iva的代表性测定中的合适的第二种酶是得自曲霉菌属种的β-半乳糖苷酶,且被用于在mps-iva酶从糖的6-位除去硫酸酯以后从它的糖苷配基释放半乳糖。这会使测定灵敏度提高约20倍。曲霉菌属酶优于例如大肠杆菌酶,因为它在ph4-5(mps-iva测定的ph)保留高活性。可替换地,可以与n-乙酰基-半乳糖胺-6-硫酸酯一起使用mps-iva底物,并且最初的mps-iv产物被在mps-vi测定中使用的相同酶(参见上面)起作用以提供糖苷配基。
实施例10
关于mps-iiia的代表性硫酸酯酶测定
用在mps-iiia的代表性测定中的合适的第二种酶是得自bakers酵母的酵母α-葡萄糖苷酶,其被用于在mps-iiia酶从葡糖胺-n-硫酸酯的氨基除去硫酸酯以后从它的糖苷配基释放葡糖胺。可替换地,在mps-iiia酶除去硫酸酯以后,可以使用乙酰辅酶a:葡糖胺n-乙酰基转移酶将游离氨基乙酰化。可以使用哺乳动物和细菌乙酰基转移酶。
实施例11
关于mps-iva的代表性测定
在该实施例中,使用细菌酶β-n-乙酰基半乳糖胺酶(β-nga)(其将β糖苷切割成n-乙酰基-半乳糖胺,当它没有在6-位硫酸化时)和人己糖胺酶a的抑制剂(z)-o-(2-乙酰氨基-2-脱氧-d-吡喃葡萄糖亚基)-氨基n-苯基氨基甲酸酯(z-pug-nac)(以阻断人己糖胺酶a对galns底物的作用),描述了mps-iva的代表性测定。
在测定中使用的galns底物具有以下结构:
在测定中使用的galns内部标准品具有以下结构:
实验1.将得自随机新生儿的干燥血斑点的3mm穿孔与0.03ml测定混合液一起温育,所述测定混合液由1mmgalns底物在含有7.5mm乙酸钡、5.0mm乙酸铈、1mmz-pug-nac和0.01mg细菌β-n-乙酰基半乳糖胺酶的50mm甲酸铵(ph4.0)中的溶液组成。所述混合物还含有0.005mm内部标准品。在37℃摇动16小时以后,将混合物用0.12ml乙腈猝灭,并将样品板离心以沉淀出沉淀物。将上清液(0.12ml)转移至新板,并加入0.12ml水。将该板放在uhplc-ms/ms仪器(具有watersacquityuhplc系统的watersxevotqms/ms)的自动采样器上。注射样品的一部分(0.01ml),并通过多重反应监测(ms/ms)来定量galns产物(底物减去硫酸酯)、内部标准品以及从galns产物和内部标准品衍生出的糖苷配基。注意到添加的内部标准品象galns产物一样通过添加细菌β-n-乙酰基半乳糖胺酶而被转化成它的糖苷配基。上面的表1显示了每种分析物的ms/ms离子峰面积。
实验1显示了使用含有在测定缓冲液中的穿孔、galns底物、内部标准品,1mmz-pug-nac和0.01mgβ-nga的血液的完整测定的结果。411,000离子峰面积的糖苷配基信号远远大于最初形成的galns产物的信号,从而表明β-nga将大部分产物转化成糖苷配基。实验1也表明,大部分内部标准品被转化成糖苷配基。使用糖苷配基和内部标准品糖苷配基的量来确定galns酶活性的量。
实验2.实验2与实验1相同,但是使用滤纸的穿孔(无血液)。大部分内部标准品被转化成糖苷配基,从而表明添加的β-nga正在工作。糖苷配基的量仅为42,000,比在有血液存在下看到的值低约10倍。该高血液/无血液比率表明,galns测定正在工作。此外,当使用得自经证实的莫基奥综合征a型患者的干燥血斑点时,糖苷配基的量类似于在没有血液存在下看到的值(45,000),从而表明所述测定正在工作以仅检测得自未受影响的个体的血液中存在的galns。
实验3.实验3显示了一种完整测定,但是不用β-nga。如预期的,大部分内部标准品未被转化成它的糖苷配基,因为z-pug-nac没有阻断来自血液的β-nag和任何己糖胺酶a。galns产物的量是121,000,且糖苷配基的量是42,900。
实验4.实验4与实验3相同,但是没有血液。实验4仅显示了galns产物的753。因而,在实验3中的121,000产物计数中的大部分是由于galns。在实验3中的产物计数不可能是由于己糖胺酶a,因为仅β-nga产生糖苷配基。在实验3和4中的糖苷配基的量是类似的,并且这代表糖苷配基存在于galns底物,其作为污染物。
实验1-4也表明了将galns产物转化成它的糖苷配基的优点;具有血液和β-nga,糖苷配基信号是411,000,并将这与galns产物的仅121,000(当它未被β-nga转化成它的糖苷配基时)进行对比。因而,获得galns测定灵敏度的4倍增加。
实验5.实验5显示了完整测定,但是缺少己糖胺酶a抑制剂z-pug-nac。因为β-nga存在,大部分内部标准品象预期的那样转化成它的糖苷配基。糖苷配基的信号是1,350,000,远大于在实验1中。这表明,己糖胺酶a如果不受抑制的话会从galns底物产生大量糖苷配基。
实验6.实验6含有血液,但是无β-nga且无z-pug-nac。象在实验5中一样,galns产物的量是110,000而不是25,100,这是由于galns对galns底物的作用。因为β-nga不存在,仅少量galns产物被转化成糖苷配基,但是糖苷配基的量保持较高在1,110,000。这是因为在没有z-pug-nac存在下血液中的己糖胺酶a对galns底物的作用。
实验7和8.实验7和8使用滤纸(无血液)。实验7包括β-nga,而实验8不包括β-nga。如预期的,在实验7中大部分内部标准品被转化成糖苷配基,但是在实验8中则不然。实验8表明,galns底物含有少量产物(820)和糖苷配基(14,500)作为污染物。这些可以通过另一轮galns底物纯化来除去。
表1中的结果清楚地表明,在干燥血斑点中内源性地存在的未受抑制的人己糖胺酶a的作用会从galns底物产生过多的糖苷配基以导致在没有己糖胺酶a抑制剂存在下的一种有用的galns测定。数据还清楚地表明,β-nga具有期望的性能:即使在有足以完全阻断己糖胺酶a的己糖胺酶a抑制剂z-pug-nac存在下,它将大部分galns产物和内部标准品转化成它们的糖苷配基。
尽管已经例证和描述了本发明的优选实施方案,应当理解,可以在其中做出不同的变化,而不脱离本发明的精神和范围。
在其中要求排他权利或特权的本发明的实施方案定义如下:
1.一种用于测定与溶酶体贮积病有关的酶的方法,其包括:
(a)使样品与第一溶液接触以提供包含一种或多种溶酶体酶的溶液;
(b)使溶液中的一种或多种溶酶体酶与要分析的每种溶酶体酶的酶底物接触,和将所述底物与所述酶一起温育足以提供包含所述样品中存在的每种溶酶体酶的酶产物的溶液的时间,
其中每种溶酶体酶的酶底物是具有碳水化合物部分和糖苷配基部分且具有下式的化合物:
其中s是这样的碳水化合物部分,其当与所述糖苷配基部分共价地偶联时提供选自以下的酶的底物:
(i)α-l-艾杜糖醛酸酶;
(ii)艾杜糖醛酸2-硫酸酯酶;
(iii)类肝素n-硫酸酯酶;
(iv)n-乙酰基-α-d-氨基葡糖苷酶;
(v)n-乙酰基半乳糖胺6-硫酸酯-硫酸酯酶;
(vi)n-乙酰基半乳糖胺4-硫酸酯-硫酸酯酶;和
(vii)β-葡糖醛酸糖苷酶;
l2是包含1-20个碳原子的连接体,其中一个或多个碳原子可以被选自n、o和s的杂原子替换,和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
l3是包含1-20个碳原子的连接体,其中一个或多个碳原子可以被选自n、o或s的杂原子替换,和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
l4是任选的,且当存在时是包含1-20个碳原子的连接体,其中一个或多个碳原子可以被选自n、o或s的杂原子替换,和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
r1是c1-c10烷基或c1-c10烷氧基;
r2在每次出现时独立地选自c1-c10烷基、c1-c10烷氧基、卤素、硝基、-c(=o)nhr或-c(=o)or,其中r是c1-c8烷基;
r3是c1-c10烷基或被取代的或未被取代的c6-c10芳基;且
n是0、1、2、3或4;和
(c)确定所述酶产物中的一种或多种的量。
2.根据实施方案1所述的方法,所述方法进一步包括使所述酶产物与糖原水解酶接触以提供第二酶产物。
3.根据实施方案1所述的方法,所述方法进一步包括添加抑制剂以阻断作用于选自以下的一种或多种酶的底物的内源性糖原水解酶活性:
(a)n-乙酰基半乳糖胺6-硫酸酯-硫酸酯酶;和
(b)n-乙酰基半乳糖胺4-硫酸酯-硫酸酯酶。
4.一种用于测定一种或多种溶酶体酶的酶活性的方法,其包括:
(a)使样品与第一溶液接触以提供包含一种或多种溶酶体酶的溶液;
(b)使溶液中的一种或多种溶酶体酶与要分析的每种溶酶体酶的酶底物接触,和将所述底物与所述酶一起温育足以提供包含存在于样品中的每种溶酶体酶的第一酶产物的溶液的时间;
(c)使所述第一酶产物遇到糖原水解酶以提供易于通过糖原水解酶实现的进一步酶促反应的每种第一酶产物的第二酶产物;和
(d)确定一种或多种第一酶产物和/或一种或多种第二酶产物的量。
5.根据实施方案4所述的方法,其中所述易于通过糖原水解酶实现的进一步酶促反应的第一酶产物通过选自以下的酶的作用来提供:
(a)艾杜糖醛酸2-硫酸酯酶;
(b)类肝素n-硫酸酯酶;
(c)n-乙酰基半乳糖胺6-硫酸酯-硫酸酯酶;和
(d)n-乙酰基半乳糖胺4-硫酸酯-硫酸酯酶。
6.根据实施方案4所述的方法,其中不易于通过糖原水解酶实现的进一步酶促反应的第一酶产物由选自以下的酶的作用提供:
(a)α-l-艾杜糖醛酸酶;
(b)n-乙酰基-α-d-氨基葡糖苷酶;和
(c)β-葡糖醛酸糖苷酶。
7.根据实施方案4所述的方法,所述方法进一步包括添加抑制剂以阻断作用于选自以下的一种或多种酶的底物的内源性糖原水解酶活性:
(a)n-乙酰基半乳糖胺6-硫酸酯-硫酸酯酶;和
(b)n-乙酰基半乳糖胺4-硫酸酯-硫酸酯酶,
其中所述抑制剂不会显著地抑制步骤(c)的糖原水解酶的活性。
8.根据实施方案1或4所述的方法,其中所述一种或多种溶酶体酶包含选自以下的酶:
(a)α-l-艾杜糖醛酸酶;
(b)艾杜糖醛酸2-硫酸酯酶;
(c)类肝素n-硫酸酯酶;
(d)n-乙酰基-α-d-氨基葡糖苷酶;
(e)n-乙酰基半乳糖胺6-硫酸酯-硫酸酯酶;
(f)n-乙酰基半乳糖胺4-硫酸酯-硫酸酯酶;和
(g)β-葡糖醛酸糖苷酶。
9.根据实施方案1或4所述的方法,所述方法进一步包括在使溶酶体酶与底物接触之前、之后或同时加入要分析的每种溶酶体酶的内部标准品。
10.根据实施方案1或4所述的方法,所述方法进一步包括在确定所述酶产物中的一种或多种的量之前猝灭酶反应。
11.根据实施方案1或4所述的方法,其中所述样品是血液或组织样品。
12.根据实施方案1或4所述的方法,其中所述样品是干燥血斑点。
13.根据实施方案2或4所述的方法,其中所述糖原水解酶选自:人己糖胺酶a、细菌n-乙酰基己糖胺酶、细菌β-n-乙酰基半乳糖胺酶、α-l-艾杜糖醛酸酶、β-半乳糖苷酶(曲霉菌属)和α-葡萄糖苷酶(酵母)。
14.根据实施方案3或7所述的方法,其中所述抑制剂选自:(z)-o-(2-乙酰氨基-2-脱氧-d-吡喃葡萄糖亚基)-氨基n-苯基氨基甲酸酯、1-脱氧野尻霉素、栗精胺、苦马豆素、打碗花精b2、isofagamine、达菲、葡糖酸肟基内酯、葡糖醛酸和它的内酯和内酰胺、扎那米韦、米格列醇、苯乙基取代的葡萄糖-和半乳糖-咪唑、n-羟基乙基脱氢野尻霉素、galnac噻唑啉和glcnac噻唑啉。
15.根据实施方案1或4所述的方法,其中确定所述酶产物的量包括质谱分析。
16.根据实施方案1或4所述的方法,其中确定所述酶产物的量包括通过质谱分析来确定每种产物与它的内部标准品的比率。
17.根据实施方案1或4所述的方法,其中确定所述酶产物的量包括串联质谱分析,其中产生产物和它们的内部标准品的母离子,分离,并进行碰撞诱导的解离以提供产物片段离子和内部标准品片段离子。
18.根据实施方案1或4所述的方法,其中确定所述酶产物的量包括对比产物片段离子和内部标准品片段离子的峰强度以计算产物的量。
19.根据实施方案1或4所述的方法,其中确定所述酶产物的量包括通过液相色谱法或通过流动注射将产物引导至质谱仪。
20.根据实施方案1或4所述的方法,其中确定所述酶产物的量包括荧光分析。
21.根据实施方案1或4所述的方法,所述方法进一步包括使用酶产物的量来确定所述样品是否来自用于治疗与一种或多种溶酶体酶缺乏有关的病症的候选物。
22.根据实施方案4所述的方法,其中所述底物具有碳水化合物部分和糖苷配基部分且具有下式:
其中
s是这样的碳水化合物部分,其当与所述糖苷配基部分共价地偶联时提供选自以下的酶的底物:
(a)α-l-艾杜糖醛酸酶;
(b)艾杜糖醛酸2-硫酸酯酶;
(c)类肝素n-硫酸酯酶;
(d)n-乙酰基-α-d-氨基葡糖苷酶;
(e)n-乙酰基半乳糖胺6-硫酸酯-硫酸酯酶;
(f)n-乙酰基半乳糖胺4-硫酸酯-硫酸酯酶;和
(g)β-葡糖醛酸糖苷酶;
l2是包含1-20个碳原子的连接体,其中一个或多个碳原子可以被选自n、o和s的杂原子替换,和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
l3是包含1-20个碳原子的连接体,其中一个或多个碳原子可以被选自n、o或s的杂原子替换,和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
l4是任选的,且当存在时是包含1-20个碳原子的连接体,其中一个或多个碳原子可以被选自n、o或s的杂原子替换,和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
r1是c1-c10烷基或c1-c10烷氧基;
r2在每次出现时独立地选自c1-c10烷基、c1-c10烷氧基、卤素、硝基、-c(=o)nhr或-c(=o)or,其中r是c1-c8烷基;
r3是c1-c10烷基或被取代的或未被取代的c6-c10芳基;且
n是0、1、2、3或4。
23.根据实施方案1或22所述的方法,其中所述底物具有下式:
24.根据实施方案1或22所述的方法,其中所述底物具有下式:
25.根据实施方案1或22所述的方法,其中所述底物具有下式:
26.根据实施方案1或22所述的方法,其中所述底物具有下式:
27.根据实施方案1或22所述的方法,其中所述底物具有下式:
28.根据实施方案1或22所述的方法,其中所述底物具有下式:
29.根据实施方案1或22所述的方法,其中所述底物具有下式:
30.根据实施方案1或22所述的方法,其中所述底物具有下式:
31.根据实施方案1或22所述的方法,其中所述底物具有下式:
32.根据实施方案1或22所述的方法,其中所述底物具有下式:
33.根据实施方案1或22所述的方法,其中所述底物具有下式:
34.根据实施方案1或22所述的方法,其中所述底物具有下式:
35.根据实施方案1或22所述的方法,其中所述底物具有下式:
36.根据实施方案1或22所述的方法,其中所述底物具有下式:
37.一种化合物,其具有碳水化合物部分和糖苷配基部分且具有下式:
其中
s是这样的碳水化合物部分,其当与所述糖苷配基部分共价地偶联时提供选自以下的酶的底物:
(a)α-l-艾杜糖醛酸酶;
(b)艾杜糖醛酸2-硫酸酯酶;
(c)类肝素n-硫酸酯酶;
(d)n-乙酰基-α-d-氨基葡糖苷酶;
(e)n-乙酰基半乳糖胺6-硫酸酯-硫酸酯酶;
(f)n-乙酰基半乳糖胺4-硫酸酯-硫酸酯酶;和
(g)β-葡糖醛酸糖苷酶;
l2是包含1-20个碳原子的连接体,其中一个或多个碳原子可以被选自n、o和s的杂原子替换,和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
l3是包含1-20个碳原子的连接体,其中一个或多个碳原子可以被选自n、o或s的杂原子替换,和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
l4是任选的,且当存在时是包含1-20个碳原子的连接体,其中一个或多个碳原子可以被选自n、o或s的杂原子替换,和/或一个或多个碳原子可以被c1-c6烷基或卤素取代;
r1是c1-c10烷基或c1-c10烷氧基;
r2在每次出现时独立地选自c1-c10烷基、c1-c10烷氧基、卤素、硝基、-c(=o)nhr或-c(=o)or,其中r是c1-c8烷基;
r3是c1-c10烷基或被取代的或未被取代的c6-c10芳基;且
n是0、1、2、3或4。
38.根据实施方案37所述的化合物,其中l2是-(ch2)n-,其中n是1-6。
39.根据实施方案37所述的化合物,其中l3是-(ch2)m-,其中m是1-12。
40.根据实施方案37所述的化合物,其中l4是-(ch2)n-,其中n是1-6。
41.根据实施方案37所述的化合物,其中l4不存在。
42.根据实施方案37所述的化合物,其中r1是c1-c5烷基。
43.根据实施方案37所述的化合物,其中r2是c1-c8烷基。
44.根据实施方案37所述的化合物,其中r3是c1-c6烷基。
45.根据实施方案37所述的化合物,其中r3是苯基。
46.具有下式的根据实施方案37所述的化合物:
47.具有下式的根据实施方案37所述的化合物:
48.具有下式的根据实施方案37所述的化合物:
49.具有下式的根据实施方案37所述的化合物:
50.具有下式的根据实施方案37所述的化合物:
51.具有下式的根据实施方案37所述的化合物:
52.具有下式的根据实施方案37所述的化合物:
53.具有下式的根据实施方案37所述的化合物:
54.具有下式的根据实施方案37所述的化合物:
55.具有下式的根据实施方案37所述的化合物:
56.具有下式的根据实施方案37所述的化合物:
57.具有下式的根据实施方案37所述的化合物:
58.具有下式的根据实施方案37所述的化合物:
59.具有下式的根据实施方案37所述的化合物:
60.一种用于测定与溶酶体贮积病有关的酶的试剂盒,其包含选自实施方案37-59的化合物中的一种或多种的底物。
61.根据实施方案60所述的试剂盒,其中所述酶选自以下的一种或多种:α-l-艾杜糖醛酸酶(mps-i)、艾杜糖醛酸-2-硫酸酯酶(mps-ii)、类肝素n-硫酸酯酶(mps-iiia)、n-乙酰基-α-d-氨基葡萄糖苷酶(mps-iiib)、n-乙酰基半乳糖胺-6-硫酸酯-硫酸酯酶(mps-iva)、n-乙酰基半乳糖胺-4-硫酸酯-硫酸酯酶(mps-vi)和β-葡糖醛酸糖苷酶(mps-vii)。
62.根据实施方案60所述的试剂盒,所述试剂盒进一步包含要测定的每种酶的内部标准品。
63.一种用于测定与溶酶体贮积病有关的酶的方法,其包括使样品与底物接触,所述底物选自实施方案37-59的化合物中的一种或多种。
64.根据实施方案63所述的方法,其中所述酶选自以下的一种或多种:α-l-艾杜糖醛酸酶(mps-i)、艾杜糖醛酸-2-硫酸酯酶(mps-ii)、类肝素n-硫酸酯酶(mps-iiia)、n-乙酰基-α-d-氨基葡萄糖苷酶(mps-iiib)、n-乙酰基半乳糖胺-6-硫酸酯-硫酸酯酶(mps-iva)、n-乙酰基半乳糖胺-4-硫酸酯-硫酸酯酶(mps-vi)和β-葡糖醛酸糖苷酶(mps-vii)。
65.根据实施方案63所述的方法,所述方法进一步包括使所述样品与要测定的每种酶的内部标准品接触。
66.一种用于测定α-l-艾杜糖醛酸酶(mps-i)的方法,所述方法包括使样品与实施方案46或47所述的化合物接触。
67.一种用于测定艾杜糖醛酸-2-硫酸酯酶(mps-ii)的方法,所述方法包括使样品与实施方案48或49所述的化合物接触。
68.一种用于测定肝素n-硫酸酯酶(mps-iiia)的方法,所述方法包括使样品与实施方案50或51所述的化合物接触。
69.一种用于测定n-乙酰基-α-d-氨基葡萄糖苷酶(mps-iiib)的方法,所述方法包括使样品与实施方案52或53所述的化合物接触。
70.一种用于测定n-乙酰基半乳糖胺-6-硫酸酯-硫酸酯酶(mps-iva)的方法,所述方法包括使样品与实施方案54或55所述的化合物接触。
71.一种用于测定n-乙酰基半乳糖胺-4-硫酸酯-硫酸酯酶(mps-vi)的方法,所述方法包括使样品与实施方案56或57所述的化合物接触。
72.一种用于测定β-葡糖醛酸糖苷酶(mps-vii)的方法,所述方法包括使样品与实施方案58或59所述的化合物接触。
- 还没有人留言评论。精彩留言会获得点赞!