一种汤料中杂环胺的检测方法与流程
本发明涉及食品检测技术领域,更具体地说,它涉及一种汤料中杂环胺的检测方法。
背景技术:
杂环胺是富含蛋白质的食品在热加工过程中,由于组织中蛋白质、氨基酸、肌酸酐和肌酸发生热分解,由碳、氮、氢组成的一类具有致癌、致突变多环芳香族化合物。
在公开号为cn107290452a的中国发明专利申请文件中公开了一种检测结合态杂环胺含量的方法,包括:对样品进行前处理;采用液相色谱质谱联用仪进行检测;所述液相色谱条件为,色谱柱是以嵌入极性基团的十八烷基硅烷键合硅胶为固定相的色谱柱;柱温为30~50℃;流动相中有机相包括乙腈,水相包括乙酸铵;流速为0.2~0.5ml/min;进样量为5~20μl;所述质谱条件为,离子源温度为100~120℃;脱溶剂气温度为350~450℃;毛细管电压为3.0~4.0kv;锥孔气流量为40~60l/h;脱溶剂气流量为600~800l/h;碰撞气流量为0.1~0.2ml/min;扫描范围为2~2000da。所述对样品进行前处理,其是向样品粉末中添加氢氧化钠溶液,混匀后加入硅藻土搅拌,再加入乙酸乙酯超声萃取,收集滤渣;装入耐压瓶内,加入盐酸,混匀后水解,活化固相萃取柱后上样洗脱,氮吹后复溶,过滤,得到处理好的样品。
上述申请文件中,提供的结合态杂环胺含量的方法,提供了17种杂环胺的定性与定量特征离子及优化的质谱参数,能够同时检测17种极性、非极性的杂环胺,且回收率十分优秀,整体速度较快,但其在对样品进行前处理的过程中,方法较为简单,并不能对样品中的干扰杂质起到良好的去除效果,而干扰杂质含量较高会导致检测结果偏差较大,不利于得到准确的测试结果,因此,需要提出一种新的方案来解决上述问题。
技术实现要素:
针对现有技术中因样品在前处理过程中残留的干扰杂质较多,而导致检测结果容易产生偏差的问题,本发明的目的一在于提供一种汤料中杂环胺的检测方法,以解决上述技术问题,其能够对干扰杂质起到良好的去除效果,并使干扰杂质的含量不易是检测结果产生较大偏差。
为实现上述目的一,本发明提供了如下技术方案:
一种汤料中杂环胺的检测方法,具体包括以下步骤:
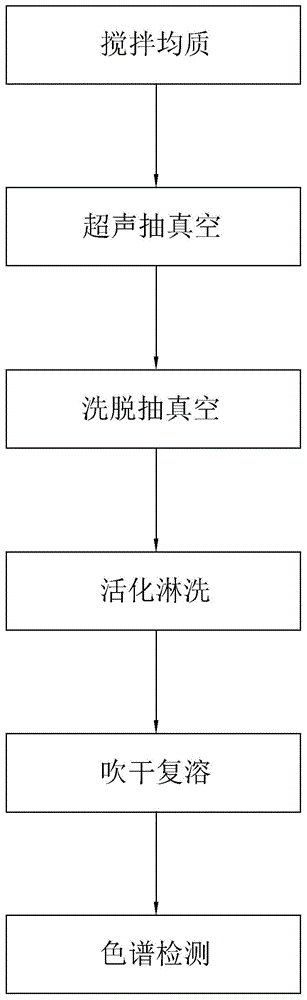
步骤一,搅拌均质:取一定量的待测样品,加入4-6倍体积的氢氧化钠溶液,搅拌均质10-20min,得到混合物;
步骤二,超声抽真空:将得到的混合物进行超声20-30min后,与2-3倍量的惰性固体物质吸附剂充分混合后填入固相萃取柱中,在﹣34-﹣32kpa压力下抽真空25-35s;
步骤三,洗脱抽真空:紧接着用二氯甲烷进行洗脱,使洗脱液自然流下,待收集的洗脱液通过固相萃取柱,在﹣34-﹣32kpa压力下抽真空2-3min,得到洗脱液;
步骤四,活化淋洗:将洗脱液预先通过用二氯甲烷活化后的固相萃取小柱,待洗脱液完全通过后依次用二氯甲烷、混合洗脱剂a、甲醇和去离子水淋洗,将杂质洗脱,再用混合洗脱剂b将杂环胺洗脱,收集得到洗脱液;
步骤五,吹干复溶:将洗脱液用50-60℃的氮气吹5-10min,再用甲醇完全复溶,得到待测样品;
步骤六,色谱检测:将待测样品用液相色谱定量检测汤料中杂环胺的含量。
通过采用上述技术方案,杂环胺属于有机碱,加入氢氧化钠进行均质,得到稳定的混合物,然后加入惰性固体物质吸附剂进行超声萃取,能够去吃一些大颗粒的干扰杂质,而再用二氯甲烷进行洗脱,能够去除微小的酸性和中性杂质。然后在活化淋洗过程中,依次用二氯甲烷、混合洗脱剂a、甲醇和去离子水淋洗,将杂质洗脱,再用混合洗脱剂b将杂环胺洗脱,能够对干扰杂质起到良好的去除效果,并使干扰杂质的含量不易是检测结果产生较大偏差,进而使含有杂环胺的待测样品在色谱检测中能够得到稳定准确的检测结果,且检测过程用时较少,整体具有良好的应用效果。
进一步优选为,所述步骤一中,氢氧化钠溶液的浓度为1.6-2.4mol/l,且均质搅拌速度为30-60rpm,温度为30-40℃。
通过采用上述技术方案,选用浓度为1.6-2.4mol/l的氢氧化钠溶液,可以使杂环胺去离子化程度增大,疏水性加强,并使杂环胺在有机溶剂中的溶解度增大,从而可提高杂环胺的回收率,保证检测结果的准确性。同时均质搅拌速度为30-60rpm,温度为30-40℃,可以保证操作过程的稳定,便于后续对干扰杂质进行去除。
进一步优选为,所述步骤二中,惰性固体物质吸附剂选用硅藻土、玻璃微珠和聚四氟乙烯中的任意一种。
通过采用上述技术方案,硅藻土、玻璃微珠和聚四氟乙烯均为良好的惰性固体吸附剂,不仅在使用过程中能够保持良好的稳定性,且能够对干扰杂质起到良好的吸附去除效果。
进一步优选为,所述步骤四中,混合洗脱剂a由0.08-0.12mol/l的盐酸溶液和甲醇按体积比为1:(1.3-1.7)混合而得。
通过采用上述技术方案,选用0.08-0.12mol/l的盐酸溶液和甲醇按体积比为1:(1.3-1.7)混合作为混合洗脱剂a,能够大大降低干扰物质的残留量,进而提高检测结果的准确性,有利于降低误差。
进一步优选为,所述步骤四中,混合洗脱剂a由0.1mol/l的盐酸溶液和甲醇按体积比为1:1.5混合而得。
通过采用上述技术方案,选用0.1mol/l的盐酸溶液和甲醇按体积比为1:1.5混合作为混合洗脱剂a,能够使混合洗脱剂a对干扰杂质起到最优的去除效果。
进一步优选为,所述步骤四中,混合洗脱剂b由12-15%体积份数的氨水和甲醇按体积比为1:(5.5-5.9)混合而得。
通过采用上述技术方案,选用12-15%体积份数的氨水和甲醇按体积比为1:(5.5-5.9)混合作为混合洗脱剂b,能够大大降低干扰物质的残留量,进而提高检测结果的准确性,有利于降低误差。
进一步优选为,所述步骤四中,混合洗脱剂b由15%体积份数的氨水和甲醇按体积比为1:5.9混合而得。
通过采用上述技术方案,选用15%体积份数的氨水和甲醇按体积比为1:5.9混合作为混合洗脱剂b,能够使混合洗脱剂b对干扰杂质起到最优的去除效果。
进一步优选为,所述步骤四中,二氯甲烷、混合洗脱剂a、甲醇和去离子水的体积比为1:(3-5):(1-3):(0.8-1.4)。
通过采用上述技术方案,选用上述体积比的二氯甲烷、混合洗脱剂a、甲醇和去离子水进行淋洗,能够对干扰杂质起到良好洗脱效果,保证检测结果的准确性。
进一步优选为,所述步骤四中,二氯甲烷、混合洗脱剂a、甲醇和去离子水的体积比为1:4:2:1.1。
通过采用上述技术方案,选用体积比为1:4:2:1.1的二氯甲烷、混合洗脱剂a、甲醇和去离子水,能够对干扰杂质起到最优的洗脱效果,有利于保证检测结果的准确性,大大降低误差值。
综上所述,与现有技术相比,本发明具有以下有益效果:
(1)加入氢氧化钠进行均质,得到稳定的混合物,然后加入惰性固体物质吸附剂进行超声萃取,再用二氯甲烷进行洗脱,然后在活化淋洗过程中,依次用二氯甲烷、混合洗脱剂a、甲醇和去离子水淋洗,将杂质洗脱,再用混合洗脱剂b将杂环胺洗脱,能够对干扰杂质起到良好的去除效果,并使干扰杂质的含量不易是检测结果产生较大偏差,进而使含有杂环胺的待测样品在色谱检测中能够得到稳定准确的检测结果;
(2)选用0.08-0.12mol/l的盐酸溶液和甲醇按体积比为1:(1.3-1.7)混合作为混合洗脱剂a;选用12-15%体积份数的氨水和甲醇按体积比为1:(5.5-5.9)混合作为混合洗脱剂b;能够大大降低干扰物质的残留量,进而提高检测结果的准确性,有利于降低误差;
(3)选用体积比为1:(3-5):(1-3):(0.8-1.4)的二氯甲烷、混合洗脱剂a、甲醇和去离子水进行淋洗操作,能够对干扰杂质起到良好洗脱效果,保证检测结果的准确性。
附图说明
图1为本发明的工艺流程图。
具体实施方式
下面结合附图和实施例,对本发明进行详细描述。
实施例1:一种汤料中杂环胺的检测方法,主要通过如下步骤制备获得:
步骤一,搅拌均质:取一定量的待测样品,加入5倍体积的氢氧化钠溶液,搅拌均质15min,而氢氧化钠溶液的浓度为2.0mol/l,且均质搅拌速度为45rpm,温度为35℃,得到混合物;
步骤二,超声抽真空:将得到的混合物进行超声25min后,与2.5倍量的硅藻土充分混合后填入固相萃取柱中,在﹣33kpa压力下抽真空30s;
步骤三,洗脱抽真空:紧接着用二氯甲烷进行洗脱,使洗脱液自然流下,待收集的洗脱液通过固相萃取柱,在﹣33kpa压力下抽真空2.5min,得到洗脱液;
步骤四,活化淋洗:将洗脱液预先通过用二氯甲烷活化后的固相萃取小柱,待洗脱液完全通过后依次用二氯甲烷、混合洗脱剂a、甲醇和去离子水淋洗,将杂质洗脱,再用混合洗脱剂b将杂环胺洗脱,收集得到洗脱液;
步骤五,吹干复溶:将洗脱液用55℃的氮气吹7.5min,再用甲醇完全复溶,得到待测样品;
步骤六,色谱检测:将待测样品用液相色谱定量检测汤料中杂环胺的含量。
注:步骤四中混合洗脱剂a由0.1mol/l的盐酸溶液和甲醇按体积比为1:1.5混合而得;混合洗脱剂b由15%体积份数的氨水和甲醇按体积比为1:5.9混合而得;二氯甲烷、混合洗脱剂a、甲醇和去离子水的体积比为1:4:2:1.1;步骤六中的色谱条件为:色谱柱tskgelods-80tm(250mm×4.6mm,5μm)(日本tosoh公司);乙腈(a)和0.01mol/l磷酸-三乙胺缓冲溶液(ph3.6)(b)为流动相进行梯队洗脱,梯度洗脱程序:0-15min,5-25%a;15-25min,25-45%a;25-30min,45-35%a;0-15min,5-25%a;31min,5%a,然后保持5min;流速为1ml/min;检测波长:253nm;室温下进样量为30μl;固相萃取小柱选购为oasismcx固相萃取小柱;固相萃取柱选购为bondelut固相萃取柱。
实施例2:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,具体包括如下步骤:
步骤一,搅拌均质:取一定量的待测样品,加入6倍体积的氢氧化钠溶液,搅拌均质10min,而氢氧化钠溶液的浓度为2.4mol/l,且均质搅拌速度为30rpm,温度为40℃,得到混合物;
步骤二,超声抽真空:将得到的混合物进行超声30min后,与2倍量的硅藻土充分混合后填入固相萃取柱中,在﹣32kpa压力下抽真空25s;
步骤三,洗脱抽真空:紧接着用二氯甲烷进行洗脱,使洗脱液自然流下,待收集的洗脱液通过固相萃取柱,在﹣32kpa压力下抽真空2min,得到洗脱液;
步骤四,活化淋洗:将洗脱液预先通过用二氯甲烷活化后的固相萃取小柱,待洗脱液完全通过后依次用二氯甲烷、混合洗脱剂a、甲醇和去离子水淋洗,将杂质洗脱,再用混合洗脱剂b将杂环胺洗脱,收集得到洗脱液;
步骤五,吹干复溶:将洗脱液用60℃的氮气吹5min,再用甲醇完全复溶,得到待测样品;
步骤六,色谱检测:将待测样品用液相色谱定量检测汤料中杂环胺的含量。
实施例3:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,具体包括如下步骤:
步骤一,搅拌均质:取一定量的待测样品,加入4倍体积的氢氧化钠溶液,搅拌均质20min,而氢氧化钠溶液的浓度为1.6mol/l,且均质搅拌速度为60rpm,温度为30℃,得到混合物;
步骤二,超声抽真空:将得到的混合物进行超声20min后,与3倍量的硅藻土充分混合后填入固相萃取柱中,在﹣34kpa压力下抽真空35s;
步骤三,洗脱抽真空:紧接着用二氯甲烷进行洗脱,使洗脱液自然流下,待收集的洗脱液通过固相萃取柱,在﹣34kpa压力下抽真空3min,得到洗脱液;
步骤四,活化淋洗:将洗脱液预先通过用二氯甲烷活化后的固相萃取小柱,待洗脱液完全通过后依次用二氯甲烷、混合洗脱剂a、甲醇和去离子水淋洗,将杂质洗脱,再用混合洗脱剂b将杂环胺洗脱,收集得到洗脱液;
步骤五,吹干复溶:将洗脱液用50℃的氮气吹10min,再用甲醇完全复溶,得到待测样品;
步骤六,色谱检测:将待测样品用液相色谱定量检测汤料中杂环胺的含量。
实施例4:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中混合洗脱剂a由0.08mol/l的盐酸溶液和甲醇按体积比为1:1.3混合而得。
实施例5:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中混合洗脱剂a由0.12mol/l的盐酸溶液和甲醇按体积比为1:1.7混合而得。
实施例6:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中混合洗脱剂b由13.5%体积份数的氨水和甲醇按体积比为1:5.7混合而得。
实施例7:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中混合洗脱剂b由12%体积份数的氨水和甲醇按体积比为1:5.5混合而得。
实施例8:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中二氯甲烷、混合洗脱剂a、甲醇和去离子水的体积比为1:3:1:0.8。
实施例9:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中二氯甲烷、混合洗脱剂a、甲醇和去离子水的体积比为1:5:3:1.4。
实施例10:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤二中,惰性固体物质吸附剂选用玻璃微珠。
实施例11:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤二中,惰性固体物质吸附剂选用聚四氟乙烯。
对比例1:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中,混合洗脱剂a由0.1mol/l的盐酸溶液和甲醇按体积比为1:1.2混合而得。
对比例2:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中,混合洗脱剂a由0.1mol/l的盐酸溶液和甲醇按体积比为1:1.8混合而得。
对比例3:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中,混合洗脱剂a由0.07mol/l的盐酸溶液和甲醇按体积比为1:1.5混合而得。
对比例4:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中,混合洗脱剂a由0.13mol/l的盐酸溶液和甲醇按体积比为1:1.5混合而得。
对比例5:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中,混合洗脱剂b由15%体积份数的氨水和甲醇按体积比为1:5.6混合而得。
对比例6:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中,混合洗脱剂b由15%体积份数的氨水和甲醇按体积比为1:6.0混合而得。
对比例7:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中,混合洗脱剂b由16%体积份数的氨水和甲醇按体积比为1:5.9混合而得。
对比例8:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中,混合洗脱剂b由11%体积份数的氨水和甲醇按体积比为1:5.9混合而得。
对比例9:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中,二氯甲烷、混合洗脱剂a、甲醇和去离子水的体积比为1:6:4:1.5。
对比例10:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中,二氯甲烷、混合洗脱剂a、甲醇和去离子水的体积比为1:2:0.8:0.7。
对比例11:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中,二氯甲烷、混合洗脱剂a、甲醇和去离子水的体积比为1:2:4:0.7。
对比例12:一种汤料中杂环胺的检测方法,与实施例1的不同之处在于,步骤四中,二氯甲烷、混合洗脱剂a、甲醇和去离子水的体积比为1:6:0.8:1.5。
性能测试
试验样品:在公开号为cn103960711a的中国发明专利中公开的牛肉汤制备方法及其牛肉汤,选用其实施例中所公开的牛肉汤配方:牛肉175-300份、牛尾骨200-400份、葱、姜、蒜各6份、大料2份、精盐2份、味精2份、香草4份、内豆蔻4份,并根据其所公开的制备方法制作出牛肉汤,取210g为试验样品。
试验方法:将210g的试验样品等质量分成21份,分别采用实施例1-11和对比例1-12所公开的汤料中杂环胺的检测方法对其进行检测,然后再根据gb5009243—2016《食品安全国家标准高温烹调食品中杂环胺类物质的测定》进行检测,计算同一试验样品两次检测的差值,并记录本发明检测方法的节省时间。
试验结果:实施例1-11和对比例1-12的测试结果如表1所示。由表1可知,由实施1-3和实施例4-11的测试结果对比可得,本发明所公开的混合洗脱剂a、混合洗脱剂b、惰性固体物质吸附剂以及二氯甲烷、混合洗脱剂a、甲醇和去离子水的体积比均适用于本检测应用,且得到的检测误差较小,并能够节省大量的时间。由对比例1-12和实施例1的测试结果对比可得,洗脱剂a的组分及配比范围、混合洗脱剂b的组分及配比范围以及二氯甲烷、混合洗脱剂a、甲醇和去离子水的配比范围均为检测最优选择范围,能够得到较为精测的检测结果。同时,表中节省时间的不同是人为操作造成的,且相差不大,在可控范围内。
表1实施例1-11和对比例1-12的测试结果
以上所述仅是本发明的优选实施方式,本发明的保护范围并不仅局限于上述实施例,凡属于本发明思路下的技术方案均属于本发明的保护范围。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理前提下的若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!