一种养殖沉积物中农兽药残留量同时测定的检测方法与流程
本发明属于农兽药检测技术领域,具体涉及一种养殖沉积物中农兽药残留量同时测定的检测方法。
背景技术:
作为现代农业生产中重要的生产投入品,农药对减少农作物病虫害及杂草的发生、提高农作物产量起到至关重要的作用。但据国家统计局年度数据,2012年以来我国农药年施用量已超过180万吨,且呈现出递增趋势。在使用过程中,由于使用方法和技术相对落后,我国农药的利用率低下,70%左右的农药未被农作物直接吸收而进入农田土壤中,并通过各类环境迁移到养殖水体中,大量附集于养殖环境沉积物,从而威胁到非目标生物及人类的健康,导致了生态系统破坏。近几十年来,抗生素使用量也呈现逐年上升趋势。一方面抗生素通过水产养殖的直接投放方式进入水体环境;另一方面借由现有水产养殖常采用塘头配套养猪或水面养鸭的养殖模式,通过畜禽粪便媒介间接进入水体环境。据文献报道,抗生素类药物在沉积物中性质稳定,半衰期较长,不易降解,可对环境造成直接污染,还可能对沉积物中微生物的耐药性产生压力,诱导具有抗生素抗性基因和耐药性的细菌的产生,从而产生严重的生态毒性。水产养殖环境中的抗生素还可通过食物链蓄积作用进入人体,对人体器官产生蓄积毒性作用,危害人体健康。因此,包括除草剂、杀虫剂在内的农药和抗生素所引起的水产养殖环境污染问题成为近年来国内外的研究热点。
现有技术中,对农兽药的残留的测定方法的研究多集中在蔬菜、水果、牛奶和动物组织中,有关沉积物中农兽药残留测定的研究鲜有报道。由于沉积物中含有大量复杂的基质,还包含大量重金属和有机质,这就给目标化合物同时提取、净化造成较大困难。而传统净化方法中主要采用的固相萃取法的步骤繁琐、耗时、费用较高。
技术实现要素:
本发明的目的在于克服现有技术缺陷,提供一种养殖沉积物中农兽药残留量同时测定的检测方法。
本发明的技术方案如下:
一种养殖沉积物中农兽药残留量同时测定的检测方法,包括如下步骤:
(1)将1.98-2.02g试样、0.14-0.16gna2edta和18-22ml的缓冲液充分混合后,再加入1.9-2.1gnacl继续混合,然后离心,获得第一上清液;该缓冲液为由乙腈和磷酸盐缓冲液组成,ph=2.9-3.1;
(2)将7-8ml上述第一上清液和复合吸附剂充分混合,然后离心,获得第二上清液;该复合吸附剂由mgso4、c18、psa和gcb以0.24-0.26∶0.04-0.06∶0.11-0.12∶0.01的质量比组成,且0.01ggcb对应7-8ml上述第一上清液;
(3)将4.5-5.5ml第二上清液于48-52℃氮吹至干,再加入0.4-0.6ml乙腈水溶液,超声溶解,再经过滤膜后,送入高效液相色谱-串联质谱仪进行检测。
在本发明的一个优选实施方案中,所述步骤(1)中的充分混合具体为:涡旋0.8-1.1min,超声4.5-5.5min,剧烈手摇1.8-2.2min。
进一步优选的,所述步骤(1)中的充分混合具体为:涡旋1min,超声5min,剧烈手摇2min。
在本发明的一个优选实施方案中,所述步骤(1)中的继续混合为手摇1min。
在本发明的一个优选实施方案中,所述缓冲液由乙腈和磷酸盐缓冲液以1∶1的体积比组成。
在本发明的一个优选实施方案中,所述mgso4、c18、psa和gcb的质量比为0.25∶0.05∶0.12∶0.01。
在本发明的一个优选实施方案中,所述步骤(3)中的乙腈水溶液中,乙腈和水的体积比为2∶8。
在本发明的一个优选实施方案中,所述高效液相色谱的检测参数为:ultimatexb-c18色谱柱,规格:150×2.1mm,5μm;柱温:40℃;流动相a:4mmol/l乙酸铵-0.1%甲酸水溶液,流动相b:0.1%甲酸甲醇,梯度洗脱;流速:0.3ml/min。
进一步优选的,所述梯度洗脱的程序如下表所示:
在本发明的一个优选实施方案中,所述质谱仪的检测参数为:电喷雾电离,正负模式切换扫描,选择反应监测模式,喷雾电压3500v,鞘气压力55arb,辅助气压力15arb,离子传输管温度320℃,雾化室加热温度220℃,碰撞气1.5mtorr。
本发明的有益效果是:
1、本发明基于高效液相色谱-串联质谱,通过优化提取溶剂、提取方式、提取时间、吸附剂组成及配比,建立了一种高通量快速定性定量分析水产养殖环境沉积物中多类农兽药残留的分析方法。
2、本发明简捷实用、准确可靠、灵敏度能达到国内外现行标准的限量要求。
3、本发明能满足日常对水产养殖环境中典型农药、兽药残留的监控要求,为水产养殖环境中典型药物残留的检测和监督管理提提供技术保障,可促进生态养殖、无公害养殖的持续发展,利于水产养殖业经济发展。
附图说明
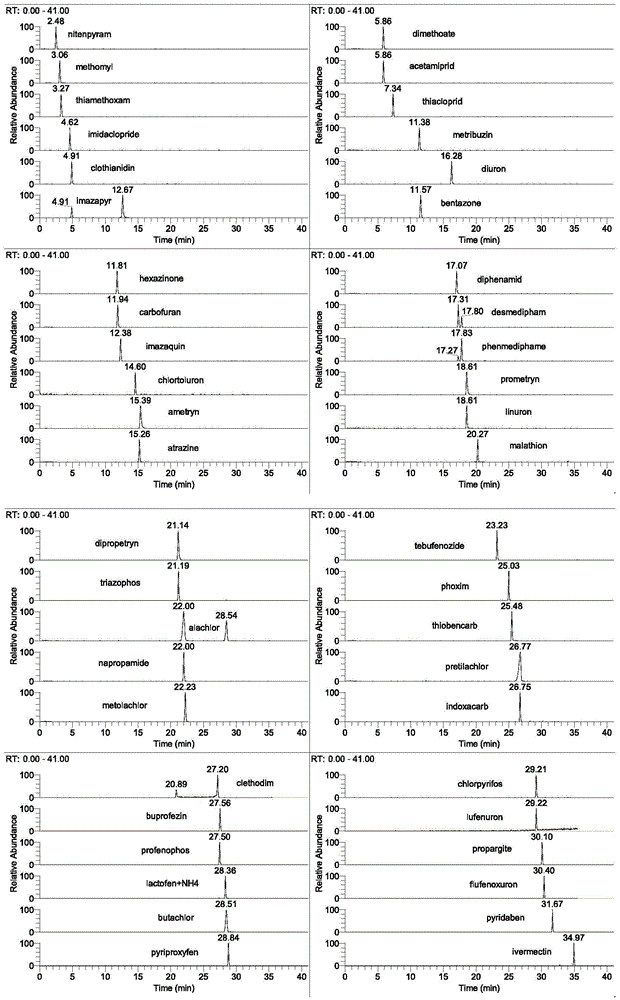
图1为本发明实施例1中的标准品选择反应监测离子流色谱图(20ng/ml)。
图2为本发明实施例1中不同提取溶剂对目标物回收率的影响图(n=3)。
图3为本发明实施例1中不同mgso4加入量对目标物回收率的影响图(n=3)。
图4为本发明实施例1中不同psa加入量对目标物回收率的影响图(n=3)。
图5为本发明实施例1中不同c18加入量对目标物回收率的影响图(n=3)。
图6为本发明实施例1中不同gcb加入量对目标物回收率的影响图(n=3)。
图7为本发明实施例1中最佳净化条件下抗生素的基质效应图。
具体实施方式
以下通过具体实施方式结合附图对本发明的技术方案进行进一步的说明和描述。
实施例1
1本实施例部分
1.1仪器与试剂
tsqquantumultra高效液相色谱-串联质谱仪(美国thermofisher公司),配电喷雾离子源;ab204-e型、pl203型电子分析天平(mettlertoledo公司);dt5-5型低速台式离心机、gt16-3高速台式离心机(北京时代北利离心机有限公司);多试管涡旋振荡器(美国vwr公司);kq600db超声波清洗器(昆山市超声仪器有限公司);hsc-24b水浴氮吹仪(天津市恒奥科技发展有限公司);milli-q型超纯水仪(美国millipore公司);fd8-6冻干机(金西盟(北京)仪器有限公司)。
灭草烟(imazapyr)、嗪草酮(metribuzin)、灭草松(bentazone)、环嗪酮(hexazinone)、灭草喹(imazaquin)、绿麦隆(chlortoluron)、莠灭净(ametryn)、莠去津(atrazine)、双苯酰草胺(diphenamid)、甜菜安(desmedipham)、甜菜宁(phenmediphame)、扑草净(prometryn)、利谷隆(linuron)、异丙净(dipropetryn)、甲草胺(alachlor)、敌草胺(napropamide)、异丙甲草胺(metolachlor)、杀草丹(thiobencarb)、丙草胺(pretilachlor)、烯草酮(clethodim)、丁草胺(butachlor)、敌草隆(diuron)、烯啶虫胺(nitenpyram)、灭多威(methomyl)、噻虫嗪(thiamethoxam)、吡虫啉(imidacloprid)、噻虫胺(clothianidin)、乐果(dimethoate)、啶虫脒(acetamiprid)、敌百虫(trichlorfon)、噻虫啉(thiacloprid)、呋喃丹(carbofuran)、马拉硫磷(malathion)、三唑磷(triazophos)、虫酰肼(tebufenozide)、辛硫磷(phoxim)、茚虫威(indoxacarb)、噻嗪酮(buprofezin)、丙溴磷(profenophos)、吡丙醚(pyriproxyfen)、毒死蜱(chlorpyrifos)、虱螨脲(lufenuron)、炔螨特(propargite)、氟虫脲(flufenoxuron)、哒螨灵(pyridaben),标准品纯度大于98%,购于美国achemtek,公司,且均为100μg/ml的标准储备液,-20℃下避光保存。再用甲醇配制1μg/ml的45种农药标准混合液,4℃下避光保存。
萘啶酸(nalidixicacid)、噁喹酸(oxolinicacid)、氟甲喹(flumequine)、磺胺吡啶(sulfapyridine)、磺胺嘧啶(sulfadiazine)、磺胺甲噁唑(sulfamethoxazole)、磺胺噻唑(sulfathiazole)、磺胺甲基嘧啶(sulfamerazine)、磺胺二甲异噁唑(sulfisoxazole)、磺胺甲噻二唑(sulfamethizol)、磺胺二甲基嘧啶(sulfamethazine)、磺胺间甲氧嘧啶(sulfamonomethoxine)、磺胺氯哒嗪(sulfachloropyridazine)、磺胺喹噁啉(sulfachinoxalin)、磺胺邻二甲氧嘧啶(sulfadoxine)、磺胺间二甲氧嘧啶(sulfadimethoxine)、盐酸克林霉素(clindamycinhydrochloride)、克拉霉素(clarithromycin)、替米考星(tilmicosin)、泰乐菌素酒石酸盐(tylosintartrate),标准品纯度大于98%,浓度为购于德国drehrenstorfer公司;交沙霉素(josamycin)标准品纯度大于99%,购于欧洲药典epcrs;三乙酸竹桃霉素(oleandomycintriacetate)、阿奇霉素二水合物(azithromycindihydrate)、红霉素(erythromycin)、罗红霉素(roxithromycin)、诺氟沙星(norfloxacin)、环丙沙星(ciprofloxacin)、恩诺沙星(enrofloxacin)、沙拉沙星(sarafloxacin)、氟罗沙星(fleroxacin),标准品纯度大于95%,购于德国drehrenstorfer公司;吉他霉素(leucomycin)标准品纯度大于92%,购于德国drehrenstorfer公司。上述标准品均用甲醇配制成100μg/ml的标准储备液,-20℃下避光保存。再用甲醇配制1μg/ml的标准混合溶液,4℃下避光保存。
甲醇、乙腈、甲酸均为色谱纯,购自美国tidea公司;乙酸铵、磷酸(h3po4)、二水合磷酸二氢钾(kh2po4·2h2o)、乙二胺四乙酸二钠(na2edta)、氯化钠(nacl)为化学纯,购自国药集团化学试剂有限公司;无水硫酸镁(mgso4)、n-丙基乙二胺(psa)、石墨化碳黑(gcb)、c18均购置天津博纳艾杰尔科技有限公司。磷酸盐缓冲液:2.72gkh2po4·2h2o与0.13mlh3po4,用超纯水定容至100ml。
沉积物采自漳州的水产养殖区,采样深度0~20cm;共10个,包括泥质、泥沙、沙质3种类型沉积物,每个样品重约500g。沉积物样品经冻干机干燥24h后研磨,过80目筛,4℃冰箱内保存。其中,沉积物样品的含水率14.1%~47.2%,有机质含量0.45%~2.11%。
1.2样品前处理
称取(2±0.02)g试样于50ml塑料离心管中,加入0.15gna2edta、20ml乙腈-磷酸盐缓冲液(1∶1,v/v),涡旋1min,超声5min,剧烈手摇2min,再加入2gnacl继续手摇1min,4000r/min离心5min;移取7~8ml上清液于15ml含有0.25gmgso4、0.05gc18、0.12gpsa、0.01ggcb的离心管中,涡旋1min,手摇1min,以3000r/min离心3min,移取5ml上清液,50℃氮吹至干。加入0.5ml水-乙腈溶液(8∶2,v/v),超声溶解,过0.22μm滤膜后供高效液相色谱-串联质谱仪测定。
1.3色谱-质谱条件
ultimatexb-c18色谱柱(150×2.1mm,5μm);柱温:40℃;流动相a:4mmol/l乙酸铵-0.1%甲酸水溶液,流动相b:0.1%甲酸甲醇;流速:0.3ml/min;梯度洗脱程序见表1。
表1梯度洗脱程序
电喷雾电离(esi),正负模式切换扫描,选择反应监测模式(srm),喷雾电压3500v,鞘气压力55arb,辅助气压力15arb,离子传输管温度320℃,雾化室加热温度220℃,碰撞气1.5mtorr;母离子、子离子和碰撞能量见表2;q1半峰宽为0.7da,q3半峰宽为0.7da。标准品选择反应监测离子流色谱图示于图1。
表2目标物质谱参数
注:*为定量碎片离子
2结果与讨论
2.1提取条件的优化
不同药物因极性和化学性质差异较大,同时提取较为困难。为了尽可能同时提取所有目标化合物,根据相似相溶原理,本实施例采用乙腈、弱酸性缓冲液作为提取溶剂。考察了1%乙酸乙腈、mcilvaine缓冲液(ph4.0)-乙腈和磷酸盐缓冲液(ph3.0)-乙腈的提取效果。结果表明(图2),磷酸盐缓冲溶液(ph3.0)-乙腈提取效率最高,可同时提取所有的目标物,且平均回收率在55.9%~112%之间。由于目标物与沉积物结合较紧密,单纯的涡旋提取无法有效提取出沉积物中的目标物。因而本实施例比较了超声辅助提取法和加速溶剂萃取法对提取效率的影响。结果表明,二者提取效率相当,但超声辅助提取法使用的溶剂较少,操作简单,有利于大批量样品的处理。同时,本实施例还考察了超声提取时间(1、2、5、7min)对目标物的提取效果。结果表明,随着提取液与目标物超声接触时间的延长,目标物提取效率随之增大。当超声时间增至5min时,目标物平均回收率趋于稳定。而当超声时间延长至7min时,目标农药和兽药回收率无影响。为了保证提取效率,同时减少杂质干扰,本实施例最终选择5min作为最终超声提取时间。此外,本实施例中edta的加入可以降低目标物与样品中金属离子的螯合,提高提取效率;而nacl的加入,则有助于分离乙腈层和水层。
现有提取方法与其它提取方法的比较的效果如下表3所示:
表3本实施例提取方法与现有技术的提取方法的对比
2.2净化条件的优化
沉积物样品中存在脂肪、色素、甾醇等杂质,因而净化的关键是吸附剂的选择,即能有效除去沉积物中杂质,又不会吸附目标物。常见的吸附材料有mgso4、psa、c18、gcb等。作为传统干燥剂,无水mgso4能有效除去有机溶剂残留的水,且吸水放热的过程可促进目标物的溶出。且mgso4粒度较小,在涡旋和振摇过程中与样品的混合更加充分,并能够与乙腈发生协同作用从而提高提取效率。psa含有两个氨基,可有效吸附沉积物中的腐蚀酸、脂肪酸、有机酸、糖类和色素。且psa属于正相吸附剂,样品含水量越少净化效果越好,因而必须协同无水mgso4才能发挥较好的净化效果。c18含有十八烷基官能团,属于非极性吸附剂,能有效除去脂肪、矿物质等非极性杂质。gcb保留特殊的层状结构,能够吸附色素、甾醇等杂质。但gcb容易吸附平面结构化合物,因此必须严控其用量。为了选择合适的吸附剂,本实施例考察了4种吸附剂量对目标物回收率的影响,设计了4个水平:mgso4(1.00、0.75、0.50、0.25g),psa(0.20、0.15、0.12、0.10g),c18(0.15、0.10、0.05、0g),gcb(0.04、0.02、0.01、0g),见图3-图6。
本实施例结果表明,添加浓度为25μg/kg水平下,单独采用mgso4、psa、c18、gcb净化,净化效果不佳;因而选用mgso4、psa、c18、gcb作为混合净化吸附剂。随着mgso4用量的增加,回收率急剧降低(图3);随着psa用量的增加,回收率先升高后降低(图4);随着ci8用量的增加,回收率先升高随后降低(图5);在保证回收率(60%~120%)的基础上,少量gcb的加入,可有效除去提取液中的色素(图6)。经优化,综合考虑净化效果和方法回收率,最终确定0.25gmgso4、0.12gpsa、0.05gc18、0.01ggcb作为净化吸附剂。
本实施例中的净化方法与其它净化方法的比较如下表4所示:
表4本实施例净化方法与现有技术的净化方法的对比
2.3基质效应
应用液相色谱-质谱测定复杂基质样品时,基质共提物对目标物的离子化具有基质增强或基质抑制效应,从而影响目标物的定量测定。基质效应(matrixeffects,me)计算公式示于式1:
me=(sm-sx)/sx×100%(1)
式中,sm和sx分别表示基质标准溶液曲线和溶剂标准溶液曲线斜率。当me<-50%或me>50%,表示存在较强基质抑制或增强效应;当-50%<me<-20%或50%≥me>20%,表示存在中等基质抑制或增强效应;当-20%≤me≤20%,表示存在较弱基质抑制或增强效应。
图7可见,在最佳提取净化条件下,7种目标物存在较强基质抑制效应,36种目标物存在中等强度的基质效应,另外28种目标物存在较弱的基质效应,不存在较强的基质效应。这说明,本实验优化的净化方法和传统spe方法一样,基本可有效消除沉积物中复杂的基质干扰物。
2.4标准曲线、线性范围、检出限和定量限
为了消除基质效应带来的定量偏差[30],实验采用基质匹配标准曲线法。移取适量混合标准溶液,用空白沉积物样品提取液分别配制成不同质量浓度基质标准溶液,目标物浓度分别为1.0,2.5,5.0,10,50,100,200μg/l。以各组分浓度与其色谱峰面积进行线性回归,呈良好线性关系,相关系数均大于0.99。以10倍信噪比(s/n)计算定量限(limitofquantitation,loq),具体数值列于表5。
2.5方法准确度和精密度
以实际采集的养殖区泥质为研究对象,进行标准添加实验,分别以低、中、高三个添加水平进行加标回收实验、每个浓度水平做6个平行实验,考察方法的准确度及精密度。表3所示,平均回收率在70.0%~114%,相对标准偏差1.5%~14.7%。30天内25μg/kg加标浓度下进行6次标准添加实验,考察方法日间精密度,相对标准偏差3.8%~11.5%(表5)。方法的精密度和准确度均能满足药物残留监测需求。选取3种类型的的沉积物——泥质、泥沙、沙质为对象,25μg/kg加标浓度下考察方法适用性,回收率70.0%~111%,相对标准偏差1.8%~13.4%(表6)。该方法适用范围广。
表5方法准确度和精密度测定结果
表6不同类型的沉积物样品加标回收率
以上所述,仅为本发明的较佳实施例而已,故不能依此限定本发明实施的范围,即依本发明专利范围及说明书内容所作的等效变化与修饰,皆应仍属本发明涵盖的范围内。
- 还没有人留言评论。精彩留言会获得点赞!