同时测定人血浆中别嘌醇和氧别嘌醇浓度的方法与流程
1.本发明涉及医药检测技术领域,具体涉及一种同时测定人血浆中别嘌醇和氧别嘌醇浓度的方法。
背景技术:
2.别嘌醇(allopurinol)是一种降尿酸药物,通过抑制黄嘌呤氧化酶来减少尿酸合成。别嘌醇与体内的次黄嘌呤为同分异构体,它及其代谢产物可抑制次黄嘌呤氧化酶,从而使尿酸生成减少降低血中尿酸浓度,减少尿酸盐在骨、关节及肾脏的沉着,有助于痛风结节及尿酸结晶的重新溶解,促使痛风结节的消散。与其他排尿酸药不同的是,别嘌醇能减少尿中尿酸的排泄量,其作用不被水杨酸盐所对抗,具有良好的市场前景。
3.现有技术(《中国药师》2009年第12卷第8期1033-1034)使用hplc-uv法测定血浆中别嘌醇及氧别嘌醇浓度,该方法需要使用大量的血浆样品,预处理加入高氯酸沉淀,涡旋混匀后高速离心并移取上清,进样体积为20μl,该方法血浆样品用量大,灵敏度低,进样量大,操作复杂,样品分析时间长约 10min,最低检测限不足,流动相中含磷酸盐对色谱柱影响大,以及样品处理程序复杂,检测时间过程不利于大批量样品检测。
4.现有技术(journal of chromatography b,941(2013)10
–
16)使用液相色谱-串联质谱法,以2,6-二氯嘌呤为内标(is)测定人血浆和尿液中别嘌醇及其活性代谢物氧嘌呤醇的浓度。别嘌醇采用正离子模式检测,分析时间约为7min。血浆中的别嘌醇浓度为0.05至5μg/ml,校准曲线呈线性。血浆中的定量下限(lloq)为0.05μg/ml。以负离子模式检测氧嘌呤醇,分析时间约为4分钟。血浆中0.05至5μg/ml (lloq,0.05μg/ml)的校准曲线呈线性。该方法通过分别采用不同的流动相和不同的ms检测模式实现别嘌醇和氧嘌呤醇的检测,没有实现同时检测且无法实现更低水平的精准定量检测,在实际运用过程中存在局限性。
技术实现要素:
5.基于现有技术的不足,本发明的目的在于建立一种lc-ms/ms法同时检测血浆中别嘌醇及氧别嘌醇的浓度的方法,克服了较低浓度水平的精准定量检测的缺陷,能满足较小给药量时生物体内的含量测定要求,检测过程中信噪比符合标准要求,无杂质峰的干扰,测定的药物的含量准确度高。
6.为了解决上述技术问题,本发明采用如下技术方案:
7.一方面,本发明提供一种同时测定人血浆中别嘌醇及氧别嘌醇浓度的方法,包括如下步骤:
8.1)标准系列工作溶液的配制:分别称取别嘌醇(bpc)和氧别嘌醇(obpc),分别经dmso溶解配制成标准对照品储备液;分别取别嘌醇标准对照品储备液和氧别嘌醇标准对照品储备液按比例混合后用50%dmso水溶液稀释,配制成混合标准曲线工作溶液;其中混合标准曲线工作溶液中,别嘌醇和氧别嘌醇的浓度比为1:10;
9.2)标准曲线血浆样本的制备:取标准系列的混合标准曲线工作溶液,稀释到空白
血浆中,配制成系列浓度的标准曲线血浆样品;系列浓度的标准曲线血浆样品中,别嘌醇和氧别嘌醇的浓度比为1:10;
10.3)标准曲线回归方程的绘制:往样品孔(96孔板)加入沉淀剂后,依次加入混合内标工作溶液、标准曲线血浆样品,封板,涡旋,离心后,进样lc-ms/ms系统,得到色谱图;根据得到的色谱图中别嘌醇色谱峰面积与内标峰面积的比值计算别嘌醇在血浆中的浓度,制定别嘌醇标准曲线回归方程;根据得到的色谱图中氧别嘌醇色谱峰面积与内标峰面积的比值计算氧别嘌醇在血浆中的浓度,制定氧别嘌醇标准曲线回归方程。
11.进一步地,所述标准对照品储备液的浓度为1mg
·
ml-1
。进一步地,所述沉淀剂为甲醇。
12.进一步地,所述混合标准曲线工作溶液中别嘌醇的浓度范围为200~20000ng/ml,氧别嘌醇的浓度范围为2000~200000ng/ml;所述混合标准曲线工作溶液中别嘌醇工作溶液的浓度范围优选为20000、17000、 10000、4000、2000、1000、400、200ng/ml,氧别嘌醇工作溶液的浓度范围优选为200000、170000、100000、 40000、20000、10000、4000、2000ng/ml。
13.进一步地,所述标准曲线血浆样品中,别嘌醇浓度范围为10~500ng/ml,优选为10、20、50、100、 200、500、850、1000ng/ml,氧别嘌醇浓度范围为100~10000ng/ml,优选为100、200、500、1000、2000、 5000、8500、10000ng/ml。
14.进一步地,所述标准曲线回归方程的获取过程如下:往样品孔加入300μl沉淀剂后,加入50μl混合内标工作溶液、50μl标准曲线血浆样品,封板,涡旋3min,离心,2490g,10min;2μl进样lc-ms/ms 系统,得到色谱图;根据得到的色谱图中别嘌醇色谱峰面积与内标峰面积的比值计算别嘌醇在血浆中的浓度,制定别嘌醇标准曲线回归方程;根据得到的色谱图中氧别嘌醇色谱峰面积与内标峰面积的比值计算氧别嘌醇在血浆中的浓度,制定氧别嘌醇标准曲线回归方程。
15.进一步地,所述混合内标工作溶液的配制过程如下:称取别嘌醇-13
c和氧别嘌醇-13
c分别于容量瓶中,经dmso溶解分别配制成1mg
·
ml-1
的别嘌醇-13
c内标对照品储备溶液和氧别嘌醇-13
c内标对照品储备溶液;
16.将别嘌醇-13
c内标对照品储备溶液用dmso稀释,得浓度为1mg
·
ml-1
的别嘌醇-13
c内标工作液;将氧别嘌醇-13
c内标对照品储备溶液用dmso稀释,得浓度为1mg
·
ml-1
的氧别嘌醇-13
c内标工作液;分别取别嘌醇-13
c(bpc is)内标工作液50μl、氧别嘌醇-13
c(obpc is)内标工作液100μl置于容量瓶中,加入50%dmso水溶液定容至刻度线,摇匀,即得浓度为别嘌醇-13
c/氧别嘌醇-13
c=500/1000ng
·
ml-1
的混合内标工作液。
17.进一步地,其中所述lc-ms/ms系统的条件如下:
18.色谱条件
19.色谱柱:phenomenex luna 3μmpfp(2)100alc column 50*4.6mm;
20.预柱:gemini c18(4*3.0mm,phenomenex,usa)
21.流动相a:0.1%甲酸水溶液;流动相b:acn
22.流速:0.650ml/min;停止时间:5.50min
23.进样量:2μl;柱温:40℃;压力:400bar
24.流动相比例设置如下:0~2min,流动相a和流动相b的体积比为98%:2%;2~3min,流动相a和流动相b的体积比为95%:5%;3~4.51min,流动相a和流动相b的体积比为
90%:10%;4.51~5.5min,流动相a和流动相b的体积比为98%:2%;
25.质谱条件
26.采用esi源,负离子mrm模式检测;
27.别嘌醇的参数设置如下:q1 mass:135,q3 mass:92,dwell:200,ce:-23,dp:-100;
28.氧别嘌醇的参数设置如下:q1 mass:151,q3 mass:108,dwell:200,ce:-30,dp:-100;
29.别嘌醇-13
c的参数设置如下:q1 mass:138,q3 mass:95,dwell:200,ce:-23,dp:-100;
30.氧别嘌醇-13
c的参数设置如下:q1 mass:154,q3 mass:111,dwell:200,ce:-30,dp:-100;
31.质谱切换阀设置如下:0~0.5mina阀;0.5~3.5min b阀;3.5min后,a阀。
32.离子源参数设置如下:cur 30;cad 7;is-4500;tem 500;gas150;gas270。
33.进一步地,所述lc-ms/ms系统检测后,别嘌醇和氧别嘌醇分别在2.54min、2.06min左右出峰,峰形良好,且人血浆中无杂质峰干扰上述化合物的测定,表明该方法具有较强的专属性。
34.进一步地,所述标准曲线的回归方程如下:
35.测得的别嘌醇在人血浆中的标准曲线回归方程为:y=0.00109x-0.00102,r为0.9992;别嘌醇的线性范围为10.000~1000.0ng/ml,别嘌醇的血药浓度的最低定量下限为10ng/ml;
36.测得的氧别嘌醇在人血浆中的标准曲线回归方程为:y=0.000566x-0.00517,r为0.9996;氧别嘌醇的线性范围为100.00~10000ng/ml,氧别嘌醇的血药浓度的最低定量下限为100ng/ml。本发明方法检测限低,灵敏度高,方法重现性好,准确可靠。
37.另一方面,本发明提供一种测定人血浆中别嘌醇及氧别嘌醇浓度的方法,具体步骤为:往样品孔加入沉淀剂后,依次加入混合内标工作溶液、标准曲线血浆样品,封板,涡旋,离心后,进样lc-ms/ms 系统,得到色谱图;根据得到的色谱图中别嘌醇色谱峰面积与内标峰面积的比值计算别嘌醇在血浆中的浓度,制定别嘌醇标准曲线回归方程;根据得到的色谱图中氧别嘌醇色谱峰面积与内标峰面积的比值计算氧别嘌醇在血浆中的浓度,制定氧别嘌醇标准曲线回归方程;
38.往样品孔加入沉淀剂后,依次加入混合内标工作溶液、含别嘌醇及氧别嘌醇的血浆样品,封板,涡旋,离心后,进样lc-ms/ms系统,得到色谱图;根据得到的色谱图的检测结果与获得的标准曲线回归方程进行计算,得到别嘌醇及氧别嘌醇浓度。
39.进一步地,所述别嘌醇及氧别嘌醇浓度获取过程为:于96孔板中加入300ul甲醇,加入50ul的isspike,精密加入50ul的别嘌醇及氧别嘌醇的血浆样品,涡旋混合3min,于4℃以2450g离心10min,2ul 进样到色谱系统进行lc-ms/ms分析,将检测结果与本技术获得的标准曲线回归方程进行计算,得到别嘌醇及氧别嘌醇浓度。
40.本发明建立lc-ms/ms法检测人血浆中的别嘌醇和氧别嘌醇浓度,能有效提高检测灵敏度,大幅减少血浆用量,节约成本;本发明选择色谱柱:phenomenex luna 3μm pfp(2)100a,规格为3μm,50
×
4.6mm;对血浆中的别嘌醇和氧别嘌醇保留效果好,峰形较好,对称且不拖尾,分离度高。对比现有技术使用的色谱柱hypersil bds c
18
柱(250mm*4.6mm,5μm)分
离耗时13min,本发明的色谱柱检测的别嘌醇和氧别嘌醇通过优化色谱梯度和流动相,将干扰峰与分析物峰成功分离,分离快速且高效,整个方法仅耗时5.5min,大大地节约成本;
41.相比现有技术的流动相,本发明选择的流动相a和流动相b,本发明流动相a为0.1%甲酸水溶液,流动相b为乙腈,相对现有技术使用的色谱柱hypersil bds c
18
柱(250mm*4.6mm,5μm)采用的流动相a为50mmol/l kh2po4;b相为甲醇,流动相成分简单,与质谱系统兼容,分析物响应良好。本发明在方法开发过程中对流速、流动相和进样量以及梯度洗脱的条件进行了优化,不仅可保证药物出峰效果和响应值,也能使药物完全洗脱干净,避免对色谱柱的污染,同时也利于回收率的提升和后续质谱检测的灵敏度的提升。
42.本发明对流动相梯度、流速等液相条件做了一系列筛选,具体如下:
43.相同流动相梯度、不同流速(0.5~0.8ml/min)检测条件下,别嘌醇、别嘌醇内标、氧别嘌醇、氧别嘌醇内标的色谱图具体比对结果发现:流速0.6ml/min和流速0.7ml/min下,别嘌醇与干扰峰次黄嘌呤分离,分离度好,但氧别嘌醇与干扰峰黄嘌呤不能分离。
44.选择流速0.7ml/min,考察相同流速,不同流动相梯度(0~3min流动相b梯度5%~20%)检测条件下,别嘌醇、别嘌醇内标、氧别嘌醇、氧别嘌醇内标的色谱图,具体比对结果发现:流速相同,流动相梯度不同,在起始梯度5%时,无法实现分离氧别嘌醇与干扰峰黄嘌呤。
45.基于流动相梯度和流速的条件摸索,发现当流动相b起始比例为5%时,不管如何调整流动相梯度、流速,氧别嘌醇和氧别嘌醇干扰峰分离度都不能完全分离,故降低流动相b起始比例为2%,但所检测的结果存在溶剂效应。
46.后续通过优化进样量、预处理沉淀剂、流速、梯度等各项参数,最终发现:本发明选择乙腈作为流动相b时,若样品前处理程序中沉淀剂选用乙腈,则会有很强的溶剂效应;若选择甲醇为沉淀剂,溶剂效应消失,适当提高流速可加快分析物出峰并改善峰形,降低进样量可有效增加分离度进而优化了本发明的液相条件和预处理过程。
47.对于质谱条件,本发明方法的响应值合理,质谱参数合适,气帘气压利于质谱液体的汽化,可持续检测。本发明采用的喷雾气压力和辅助加热气压力均设置合理,不容易损坏仪器,可持续长期检测,方法耐用性更优。通过质谱仪很强的组分鉴别能力,能够有效分离和鉴别血浆中别嘌醇和氧别嘌醇,并进行定量检测。
48.本发明方法生物样本用量小,每次血浆样品测试仅需50μl,进样量仅需2μl,可准确测定出化合物。
49.本发明方法检测得到的lc-ms图和空白基质图结合发现,能够有效地将别嘌醇、氧别嘌醇以及体内的同分异构体次黄嘌呤分离开。
50.相比现有液相检测方法,本发明方法同时检测血浆中别嘌醇和氧别嘌醇两个化合物浓度,且无基质效应和干扰,检测下限能满足较小给药量时生物体内的含量测定要求,检测过程中信噪比符合标准要求,无杂质峰的干扰,测定的药物的含量准确度高;样本前处理简单,使用的生物样本量少;采用的液相流动相简单,不含磷酸盐,能有效保护色谱柱。
51.与现有技术相比,本发明具有如下的有益效果:
52.本发明建立lc-ms/ms法同时检测血浆中别嘌醇及氧别嘌醇两个化合物的浓度,血浆样品用量为50μl,采用的样品预处理方法以甲醇沉淀蛋白为主,并不需要移取上清,操作简单快速,通过优化色谱条件能有效的将别嘌醇与体内的同分异构体次黄嘌呤分开,无基
质效应和干扰,进样量仅需2μl,灵敏度高,氧别嘌醇和别嘌醇的最低检测浓度分别为100.00ng/ml和10.000ng/ml,样品分析时间短,成本低,适合大批量样品分析。
53.本发明提供了一种预处理方法简便的测定人血浆中的别嘌醇及氧别嘌醇浓度的方法,在本实验所采用的色谱条件下,别嘌醇保留时间为2.54min左右,氧别嘌醇保留时间为2.06min左右,峰形良好,无杂峰干扰测定,基线平稳;本方法具有较高的特异性,能准确测定血浆中的别嘌醇及氧别嘌醇的浓度,灵敏度较高,血浆最低定量限分别为别嘌醇:10.000ng/ml、氧别嘌醇:100.00ng/ml;本发明方法快速、准确、灵敏度高、操作简便,为别嘌醇及氧别嘌醇的血药浓度测定提供依据。本方法别嘌醇及氧别嘌醇的血浆标准曲线线性范围分别为别嘌醇:10.000~1000.0ng/ml、氧别嘌醇:100.00~10000ng/ml,批内和批间精密度rsd均小于
±
15%。本发明方法重现性好,准确度高,不受基质、高血脂基质和溶血基质的干扰,无潜在药物干扰,不同人群的血浆对本发明方法无干扰现象。
54.本发明方法能够满足人体药代动力学要求,可应用于临床药代动力学研究。
附图说明
55.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
56.图1lc-ms/ms法测得的人血浆中别嘌醇的标准曲线图;
57.图2lc-ms/ms法测得的人血浆中氧别嘌醇的标准曲线图;
58.图3lc-ms/ms法测得的人血浆中别嘌醇(a)和别嘌醇-13
c(b)的液质图;其中,2.52min的峰是别嘌醇的分析物峰,1.91min的峰就是次黄嘌呤的干扰峰;
59.图4lc-ms/ms法测得的人血浆中氧别嘌醇(a)和氧别嘌醇-13
c(b)的液质图;其中,2.31min的峰是氧别嘌醇的分析物峰,2.07min的峰就是黄嘌呤的干扰峰;
60.图5别嘌醇负离子多反应检测扫描质谱图;
61.图6别嘌醇-13
c负离子多反应检测扫描质谱图;
62.图7氧别嘌醇负离子多反应检测扫描质谱图;
63.图8氧别嘌醇-13
c负离子多反应检测扫描质谱图;
64.图中,analyte conc./is conc.分析物浓度/标准物浓度;analyte area/is area分析物面积/标准物面积; time时间;intensity强度;counts vs.mass-to-charge计数与电荷质量
具体实施方式
65.下面结合具体实施例,进一步阐述本发明。这些实施例仅用于说明本发明而不用于限制本发明的范围。
66.1.主要仪器
67.ab sciex 6500型三重四极杆液质联用仪(ab sciex公司),配有电喷雾电离源及analyst (version1.6.3)数据处理系统,配有exionlc液相色谱仪(自动进样器、柱温箱等)(ab sciex公司);mettler toledo xpr2/a型百万分之一天平(mettler,瑞士)。
68.2.色谱条件
69.色谱柱:phenomenex luna 3μm pfp(2)100alc column 50*4.6mm;预柱:gemini c18(4*3.0mm, phenomenex,usa);流动相a:0.1%甲酸水溶液;流动相b:acn;流速:0.650ml/min;停止时间: 5.50min;进样量:2μl;柱温:40℃;流动相比例设置如下表1:
[0070] timeabmax pressμre10.00min98.0%2.0%400.00bar22.00min95.0%5.0%400.00bar33.00min10.0%90.0%400.00bar44.50min10.0%90.0%400.00bar54.51min98.0%2.0%400.00bar65.50min98.0%2.0%400.00bar
[0071]
质谱条件
[0072]
采用esi源,负离子mrm模式检测。具体参数如下表2:
[0073]
nameistdq1q3dwellcedpbpcno13592200-23-100obpcno151108200-30-100bpcisyes13895200-23-100obpcisyes154111200-30-100
[0074]
质谱切换阀设置如下表3:
[0075]
indexstarttime(min)position10a20.5b33.5a
[0076]
离子源参数设置如下表4:
[0077]
parametervalue(-)parametervalue(-)cur30tem500cad7gas150is-4500gas270
[0078]
4.内标:别嘌醇-13
c,氧别嘌醇-13c[0079]
6.数据处理
[0080]
色谱保留时间及色谱峰面积由agilentmass hunterworkstation software采集处理与定量分析,采用 agilentecm网络系统。以分析物峰面积与内标峰面积比对标准曲线中分析物的理论浓度进行线性最小二乘法回归计算,以所得回归方程计算样品中分析物的实测浓度。
[0081]
7.样品进行检测前的预处理方法:
[0082]
取全血,加入抗凝剂肝素钠,通过离心后制得空白血浆,冷藏用于进行样品稀释或配制,使用前解冻。
[0083]
往样品孔中(96孔板)加入300μl沉淀剂甲醇。
[0084]
加入50μl 50%dmso水至double blank、carryover、rb孔,加入50μl内标(is spike, 500.00/5000.0ng/ml)至sst样品、blank样品、标准曲线血浆样品、qc血浆样品对应样品孔中。
[0085]
分别移取空白血浆样品(doubleblank、blank、carryover)、超纯水(rb)、sst样品、标准曲线血浆样品(std1~8)、qc血浆样品各50μl至对应样品孔中。
[0086]
封板,涡旋3min。
[0087]
离心,2450g,10min。
[0088]
2μl进样到色谱系统分析。
[0089]
备注:doubleblank为双空白样品、carryover为残留考察用双空白样品、blank为只加内标的单空白样品、reagent and materials blank为试剂耗材验收空白样品。
[0090]
实施例1
[0091]
1.溶液配制
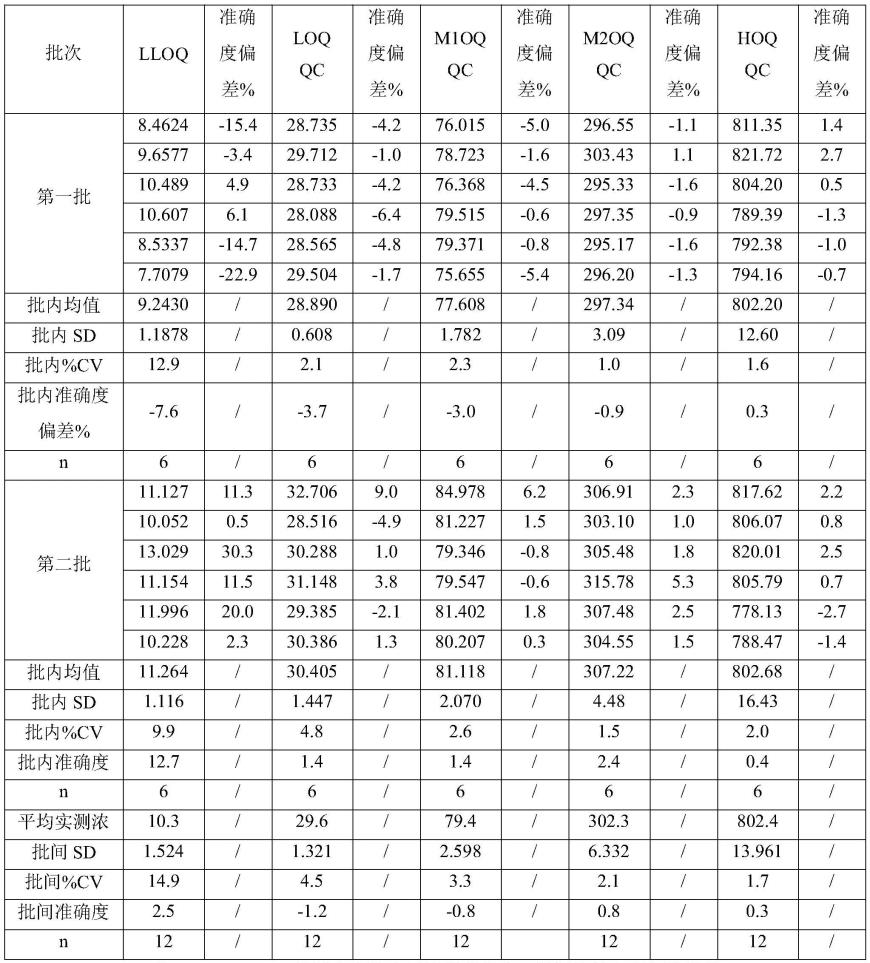
[0092]
流动相a:(0.1%甲酸溶液)量取1l超纯水,加入1ml甲酸,混匀。
[0093]
流动相b:(乙腈acn)量取1l乙腈至适当的溶剂瓶中。
[0094]
稀释溶液(dmso∶水,50∶50,v/v):分别取50ml dmso和50ml超纯水转移至适当的溶剂瓶中,混匀。
[0095]
洗针溶液配制(dmso∶水,50∶50,v/v):取500ml dmso与500ml纯化水至适当的溶剂瓶中,混匀。
[0096]
沉淀剂(甲醇):量取100ml甲醇转移至适当的溶剂瓶中。
[0097]
2.标准溶液配制:
[0098]
标准对照品储备液的配制
[0099]
别嘌醇标准对照品储备液的配制(bpc std stock,1mg/ml)
[0100]
称取一定量别嘌醇对照品,置于铝箔中,记录重量,将铝箔纸置于棕色广口玻璃瓶中,根据实际称量值及别嘌醇含量计算分析物浓度达到1.0000mg/ml所需要补加稀释液dmso的体积,加入适量dmso,最终配制成的bpc std stock浓度为1.0000mg/ml,摇匀。置于-20℃条件下保存。
[0101]
氧别嘌醇标准对照品储备液的配制(obpc std stock,1mg/ml)
[0102]
称取一定量氧嘌呤醇对照品,置于铝箔中,记录重量,将铝箔纸置于棕色广口玻璃瓶中,根据实际称量值及别嘌醇含量计算分析物浓度达到1.0000mg/ml所需要补加稀释液dmso的体积,加入适量 dmso,最终配制成的obpc std stock浓度为1.0000mg/ml,摇匀。置于-20℃条件下保存。
[0103]
标准系列工作溶液的配制:
[0104]
std spike8:取bpc std stock源溶液体积40μl,obpc std stock源溶液体积400μl,于2ml容量瓶中混合后,添加50%dmso1560μl进行定容,获得溶液中bpc/obpc浓度20000/200000ng/ml。
[0105]
std spike7:取bpc std stock源溶液体积34μl,obpc std stock源溶液体积340μl,于2ml容量瓶中混合后,添加50%dmso1626μl进行定容,获得溶液中bpc/obpc浓度17000/170000ng/ml。
[0106]
std spike6:取std spike 8源溶液体积500μl,添加50%dmso500μl进行定容,于
1ml容量瓶中混合后,获得溶液中bpc/obpc浓度10000/100000ng/ml。
[0107]
std spike5:取std spike 8源溶液体积200μl,添加50%dmso800μl进行定容,于1ml容量瓶中混合后,获得溶液中bpc/obpc浓度4000.0/40000ng/ml。
[0108]
std spike4:取std spike 8源溶液体积100μl,添加50%dmso900μl进行定容,于1ml容量瓶中混合后,获得溶液中bpc/obpc浓度2000.0/20000ng/ml。
[0109]
std spike3:取std spike 6源溶液体积100μl,添加50%dmso900μl进行定容,于1ml容量瓶中混合后,获得溶液中bpc/obpc浓度1000.0/10000ng/ml。
[0110]
std spike2:取std spike 5源溶液体积100μl,添加50%dmso900μl进行定容,于1ml容量瓶中混合后,获得溶液中bpc/obpc浓度400.0/4000ng/ml。
[0111]
std spike1:取std spike 4源溶液体积100μl,添加50%dmso900μl进行定容,于1ml容量瓶中混合后,获得溶液中bpc/obpc浓度200.0/2000ng/ml。
[0112]
注:工作液均存于-20℃条件保存。
[0113]
标准曲线血浆样品的配制
[0114]
sst:取std spike1源溶液体积50μl,于1ml容量瓶中,添加空白基质950μl进行定容,获得基质中bpc/obpc浓度10.000/100.00ng/ml。
[0115]
std 1:取std spike1源溶液体积50μl,于1ml容量瓶中,添加空白基质950μl进行定容,获得基质中bpc/obpc浓度10.000/100.00ng/ml。
[0116]
std 2:取std spike2源溶液体积50μl,于1ml容量瓶中,添加空白基质950μl进行定容,获得基质中bpc/obpc浓度20.000/200.00ng/ml。
[0117]
std 3:取std spike3源溶液体积50μl,于1ml容量瓶中,添加空白基质950μl进行定容,获得基质中bpc/obpc浓度50.000/500.00ng/ml。
[0118]
std 4:取std spike4源溶液体积50μl,于1ml容量瓶中,添加空白基质950μl进行定容,获得基质中bpc/obpc浓度100.000/1000.00ng/ml。
[0119]
std 5:取std spike5源溶液体积50μl,于1ml容量瓶中,添加空白基质950μl进行定容,获得基质中bpc/obpc浓度200.000/2000.00ng/ml。
[0120]
std 6:取std spike6源溶液体积50μl,于1ml容量瓶中,添加空白基质950μl进行定容,获得基质中bpc/obpc浓度500.000/5000.00ng/ml。
[0121]
std7:取std spike7源溶液体积50μl,于1ml容量瓶中,添加空白基质950μl进行定容,获得基质中bpc/obpc浓度850.000/8500.00ng/ml。
[0122]
std 8:取std spike8源溶液体积50μl,于1ml容量瓶中,添加空白基质950μl进行定容,获得基质中bpc/obpc浓度1000.000/10000.00ng/ml。
[0123]
注:每个浓度级别分别以65μl进行分装,分装于1.5ml离心管中,置于-70℃条件下保存,直到分析测定。
[0124]
3.内标标准溶液配制
[0125]
内标储备溶液配制
[0126]
bpc-13
c,15n2对照品储备溶液配制(bpc is stock,1.0000mg/ml)
[0127]
直接加入971μldmso,溶解规格为1mg的对照品,摇匀,即得浓度为1.0000mg/ml的此对照品储备溶液,置于-20℃条件下保存。
[0128]
obpc-13
c,15n2对照品储备溶液配制(obpc is stock,1.0000mg/ml)
[0129]
直接加入971μldmso,溶解规格为1mg的对照品,摇匀,即得浓度为1.0000mg/ml的此对照品储备溶液,置于-20℃条件下保存。
[0130]
混合内标工作溶液(is spike)配制
[0131]
取bpc-13
c is stock 50μl与obpc-13
c is stock 100μl至100ml容量瓶中,用50%dmso溶液稀释至刻度,混匀得到is spike(500/1000ng/ml)。
[0132]
4.质控标准溶液配制
[0133]
质控对照品储备液的配制:
[0134]
别嘌醇质控对照品储备液的配制(bpc qc stock,1.0000mg/ml)
[0135]
称取一定量别嘌醇对照品,置于铝箔中,记录重量,将铝箔置于棕色广口玻璃瓶中,根据实际称量值及别嘌醇含量计算分析物浓度达到1.0000mg/ml所需要补加稀释液dmso的体积,加入适量dmso,最终配制成的bpc qc stock浓度为1.0000mg/ml,摇匀。置于-20℃条件下保存。
[0136]
氧嘌呤醇质控对照品储备液的配制(obpc qc stock,1.0000mg/ml)
[0137]
称取一定量氧嘌呤醇对照品,置于铝箔中,记录重量,将铝箔置于棕色广口玻璃瓶中,根据实际称量值及氧嘌呤醇含量计算分析物浓度达到1.0000mg/ml所需要补加稀释液dmso的体积,加入适量 dmso,最终配制成的obpc qc stock浓度为1.0000mg/ml,摇匀。置于-20℃条件下保存。
[0138]
质控工作溶液的配制:
[0139]
qc spike6:取bpc qc stock源溶液体积80μl,obpc qc stock源溶液体积800μl,于1ml容量瓶中混合后,添加50%dmso120μl进行定容,获得溶液中bpc/obpc浓度80000/800000ng/ml。
[0140]
qc spike5:取qc spike 6源溶液体积200μl,于1ml容量瓶中后,添加50%dmso800μl进行定容,获得溶液中bpc/obpc浓度16000.0/60000ng/ml。
[0141]
qc spike4:取qc spike 6源溶液体积72μl,添加50%dmso888μl进行定容0.96ml,获得溶液中 bpc/obpc浓度6000.0/60000ng/ml。
[0142]
qc spike3:取qc spike 5源溶液体积100μl,于1ml容量瓶中后,添加50%dmso900μl进行定容,获得溶液中bpc/obpc浓度1600.0/16000ng/ml。
[0143]
qc spike2:取qc spike 4源溶液体积100μl,于1ml容量瓶中后,添加50%dmso900μl进行定容,获得溶液中bpc/obpc浓度600.0/6000ng/ml。
[0144]
qc spike1:取qc spike3源溶液体积125μl,于1ml容量瓶中后,添加50%dmso875μl进行定容,获得溶液中bpc/obpc浓度200.0/2000ng/ml。
[0145]
质控血浆样品的配制:
[0146]
aql qc:取qc spike6源溶液体积20μl,添加空白基质380μl定容至0.4ml,获得基质中bpc/obpc 浓度4000.000/40000.00ng/ml。
[0147]
lloq:取qc spikel源溶液体积20μl,添加空白基质380μl定容至0.4ml,获得基质中bpc/obpc 浓度10.000/100.00ng/ml。
[0148]
lqc:取qc spike2源溶液体积20μl,添加空白基质380μl定容至0.4ml,获得基质中bpc/obpc 浓度30.000/300.00ng/ml。
[0149]
m1qc:取qc spike3源溶液体积20μl,添加空白基质380μl定容至0.4ml,获得基质
中bpc/obpc 浓度80.000/800.00ng/ml。
[0150]
m1qc:取qc spike4源溶液体积20μl,添加空白基质380μl定容至0.4ml,获得基质中bpc/obpc 浓度300.000/3000.00ng/ml。
[0151]
hqc:取qc spike5源溶液体积20μl,添加空白基质380μl定容至0.4ml,获得基质中bpc/obpc 浓度800.000/8000.00ng/ml。
[0152]
注:每个浓度级配制体积可根据比例调整。
[0153]
5.方法学验证
[0154]
5.1线性范围和定量下限
[0155]
取标准系列工作溶液各10μl,稀释到空白血浆中使得总体积为0.2ml,配制的标准曲线血浆样品中别嘌醇及氧别嘌醇浓度值为:10.000/100.00ng/ml。
[0156]
往样品孔中(96孔板)加入300μl沉淀剂甲醇。
[0157]
加入50μl 50%dmso水至double blank、carryover、rb孔,加入50μl内标(is spike, 500.00/5000.0ng/ml)至sst样品、blank样品、标准曲线血浆样品、qc血浆样品对应样品孔中。
[0158]
分别移取空白血浆样品(doubleblank、blank、carryover)、超纯水(rb)、sst样品、标准曲线血浆样品(std1~8)、qc血浆样品各50μl至对应样品孔中。
[0159]
封板,涡旋3min。
[0160]
离心,2450g,10min。
[0161]
2μl进样到色谱系统分析。
[0162]
结论:所得标准曲线样品的线性曲线图如图1~2所示,其中lc-ms/ms法测得的别嘌醇及氧别嘌醇在人血浆中的标准曲线方程分别为y=0.00109x+-0.00102(别嘌醇)、y=0.000566x+-0.00517(氧别嘌醇), r分别为0.9992(别嘌醇)、0.9996(氧别嘌醇);别嘌醇的线性范围为10.000~1000.0ng/ml,氧别嘌醇的线性范围为100.00~10000ng/ml,别嘌醇的血药浓度的最低定量下限为10.000ng/ml,氧别嘌醇的血药浓度的最低定量下限为100.00ng/ml,在线性范围内线性关系良好。
[0163]
5.2精密度与准确度
[0164]
取四个不同浓度的质控血浆样品,别嘌醇及氧别嘌醇质控血浆样品液的浓度分别为800.00/8000.0ng/ml、300.00/3000.0ng/ml、80.000/800.00ng/ml、30.000/300.00ng/ml、10.000/100.00ng/ml;分别对应为hoq qc、m2oq qc、m1oq qc、loq qc、lloq浓度,并将其进行检测。作为批内及批间准确度和精密度的考察。每一个浓度平行制备6份,至少测试三次。采用平均值来评估批间准确度和精密度。结果如表5-6:
[0165]
表5:lc-ms/ms法测定血浆中别嘌醇批内、批间的精密度和准确度
[0166][0167]
表6:lc-ms/ms法测定血浆中氧别嘌醇批内、批间的精密度和准确度
[0168]
[0169][0170]
结果表明:别嘌醇及氧别嘌醇的血浆样品批内、批间精密度、准确度偏差均小于15%。低、中、高浓度水平标准血浆样品测得值应为标示值的15%范围内,批内与批间精密度(rsd)小于15%。说明本发明方法对血浆中的别嘌醇及氧别嘌醇的检测具有良好的精密度和准确度。
[0171]
5.3系统适用性:
[0172]
浓度别嘌醇为10.000ng/ml,氧别嘌醇为100.00ng/ml;2μl进样检测,重复分析5次;
[0173]
分别记录待测物与内标的色谱峰面积值及保留时间,并计算待测物与内标的色谱峰面积比值及色谱峰保留时间的变异系数。
[0174]
结论:待测物信噪比(s/n不小于5),5次重复分析所得待测物与内标色谱峰面积比值的变异系数应小于5%;5次重复分析所得待测物与内标色谱峰保留时间的变异系数应小于15.0%。待测保留时间变异系数别嘌醇:0.4%、氧别嘌醇:0.4%,内标保留时间变异系数别嘌醇:0.3%、氧别嘌醇:0.4%,面积比的变异系数别嘌醇:7.7%、氧别嘌醇:3.3%;说明本发明方法对血浆中药物的检测方法适用测定别嘌醇及氧别嘌醇。
[0175]
5.4选择性
[0176]
选择性是指色谱方法测定分析物时,区分生物基质中干扰的能力。方法验证之前,需要至少筛选6 个不同批次的空白基质用于方法验证。
[0177]
取6个中国人群不同来源的空白血浆50μl,经预处理为sele blank样品后,2μl进样到色谱系统分析。
[0178]
取50μl 6份来自不同来源的空白血浆,配制成的别嘌醇质控血浆样品液的浓度为10.000ng/ml,经预处理后,2μl进样到色谱系统分析;
[0179]
取50μl 6份来自不同来源的空白血浆,配制成氧别嘌醇质控血浆样品液的溶液为100.00ng/ml,经预处理后,2μl进样到色谱系统分析;
[0180]
分别记录各份空白样品及定量下限浓度水平的标准血浆样品的结果。
[0181]
结论:至少6份不同来源空白基质样品中的分析物保留时间处色谱峰峰面积低于相对应的不同来源空白基质lloq浓度待测物峰面积的20.0%,且内标保留时间处内标峰面积低于相对应的不同来源空白基质lloq浓度内标峰而积的5.0%。6份不同来源空白基质配制的lloq浓度测得值在理论值的 80.0%~120.0%范围内。本发明方法针对不同来源空白基质样品中的分析物的干扰范围为别嘌醇: 0.0~12.7%、氧别嘌醇:0.0~1.8%,内标干扰响应值为别嘌醇:0.0%、氧别嘌醇:0.0%。可见,不同人体的空白血浆对别嘌醇及氧别嘌醇的检测结果没有造成干扰,该方法可以用于检测不同人体血浆中别嘌醇及氧别嘌醇,不同人群的血浆对本发明检测方法无影响。
[0182]
5.5回收率
[0183]
分析物的提取回收率
[0184]
分析物回收样品
[0185]
分别取浓度为800.00/8000.0、300.00/3000.0、30.000/300.00ng/ml别嘌醇/氧别嘌醇质控血浆样品液,平行制备6份;
[0186]
加入混合内标工作溶液,沉淀后,涡流混合离心后测定。
[0187]
回收率参比样品:
[0188]
提取过程与分析物回收样品操作相同,但是混合内标工作溶液在沉淀后加入,再取上清涡流后进行测定。
[0189]
内标物的提取回收率
[0190]
内标的提取回收率样品
[0191]
由空白血浆样品平行制备6份,加入混合内标工作溶液,沉淀后,取合适体积的上清溶液,加入与 moq qc质控样品浓度相当的标准品溶液。
[0192]
内标的回收率参比样品:
[0193]
提取过程与内标的提取回收率样品的提取操作相同,但是moq qc质控样品和内标溶液均在沉淀后加入。
[0194]
结论:1)本发明方法的待测物别嘌醇及氧别嘌醇在高、中、低三个浓度范围内的提取回收率分别为别嘌醇:107.4%、109.4%、111.5%,氧别嘌醇:107.9%、109.5%、110.6%;对应的变异系数cv值为别嘌醇:0.9%、1.9%、6.9%,氧别嘌醇:2.0%、1.8%、2.9%;总回收率为别嘌醇:109.4%,氧别嘌醇:109.3%;总体回收率变异cv值为别嘌醇:4.3%,氧别嘌醇:2.4%。
[0195]
2)本发明方法的内标物的提取回收率为别嘌醇is:91.4%,obpc is:91.3%;回收
率cv值为别嘌醇is:1.9%,氧别嘌醇is:1.8%。
[0196]
5.6基质效应
[0197]
1)基质效应系指生物基质中存在的组分对分析物的离子化所产生的抑制或增强作用。分别取6个不同批次的空白人血浆,每个批次配制一份,经前处理后分别加入与处理后的低浓度和高浓度质控样品浓度相当的标准品溶液及内标溶液进行分析。
[0198]
无基质存在的与低浓度和高浓度质控样品浓度相当的纯溶液,平行6份,进行分析。
[0199]
2)溶血基质的配制:取100μl空白全血,将其超声破坏血细胞,取其20μl加入980μl正常空白血浆,混匀,即为2%经超声破坏的溶血血浆,视为严重溶血。
[0200]
使用模拟的溶血血浆制备待测物两个浓度水平(loq qc、hoq qc)样品各6份,并将其处理后,进样到色谱系统分析。
[0201]
3)使用高血脂血浆样品制备待测物低、高浓度水平(loq qc、hoq qc)样品各6份,并将其处理后,进样到色谱系统分析。
[0202]
结论:经内标归一化的基质因子的变异系数cv%为别嘌醇:0.9~4.1%,氧别嘌醇:1.7~2.3%;溶血基质效应的平均准确度偏差范围为别嘌醇:-0.1~-1-3%,氧别嘌醇:-2.7~-0.8%;高血脂基质效应的平均准确度偏差范围为别嘌醇:1.2~2.9%,氧别嘌醇:3.2~-3.6%;说明本发明方法没有明显的基质效应,也没有明显的溶血和高血脂基质效应。
[0203]
5.7稳定性
[0204]
将待测物低、高(loq qc、hoq qc)两个浓度的人血浆质控样品,在如下条件下进行稳定性考察:
[0205]
室温放置3h稳定。
[0206]
全血稳定性:分别考察加入含低、高(loq qc、hoq qc)两个浓度水平分析物在全血中冰水浴下放置(0h)、(0.5~2h)峰而积之比的变化,在放置相应考察时间点后,将全血样品离心分离血浆,加入内标按照相应血浆样品处理操作方法处理,每个浓度水平平行6份。
[0207]
通过稳定性考察后的样品与现配制的样品同时检测,每个浓度对比结果如下:
[0208]
表7室温放置3h稳定-别嘌醇短期稳定性结果:
[0209][0210]
[0211]
续表7室温放置3h稳定-氧别嘌醇短期稳定性结果:
[0212][0213]
表8别嘌醇全血稳定性结果:
[0214][0215]
续表8氧别嘌醇全血稳定性结果:
[0216]
[0217][0218]
本发明的方法检测的血浆中各药物的浓度在上述考察条件下得出的cv值小于15%,提示在上述考察条件下本发明方法对全血和血浆中别嘌醇及氧别嘌醇的检测具有较强的稳定性。
[0219]
5.8特异性
[0220]
特异性是生物分析方法检测和区分待测物与其他物质的能力,包括相关物质(例如与待测物结构相似的物质、代谢物、异构体、杂质、制备过程中形成的降解产物等),当生物分析方法中有超过一个分析物被测定并且已知分析物能够转化成另一个分析物时,需要考察分析物之间的相互转化及干扰情况。
[0221]
分析物对内标的干扰
[0222]
为评估分析物对内标的干扰,分别平行制备、处理并分析6份仅含分析物的且不加内标的定量上限浓度样品,并将其处理后,进样到色谱系统分析。
[0223]
内标对分析物的干扰
[0224]
为评估分析物对内标的干扰,分别平行制备、处理并分析六份仅含分析物且不加内标的定量上限浓度样品,并将其处理后,进样到色谱系统分析。
[0225]
潜在药物干扰
[0226]
同时检测多个化合物时,应进行潜在药物的考察。考察方法为配制并处理含有uloq浓度联合用药的低、高浓度水平的待分析物质控样品,每个浓度水平平行6份,对于因对照品不纯产生干扰的情况下,为避免两个待测物数据测量的准确性,避免对照品相互干扰带来的检测影响,潜在药物的干扰的考察方法为在空白基质中加入含联合用药标准曲线最高浓度工作液作为潜在药物干扰的空白基质(加入内标),该基质按照相应处理步骤平行处理三份计算含联合用药标准曲线最高浓度的空白基质样品中响应待测物干扰组分的浓度。
[0227]
结论:考察分析物对内标的干扰时,单个样品内标保留时间处的峰面积不大于符合接受标准的当批次的标准曲线样品和质控样品的内标的平均峰面积的5.0%,别嘌醇/氧别嘌醇分析物干扰为 0.0%/0.0%~0.1%;考察内标对分析物的干扰时,单个分析物保留时间处的峰面积不大于分析物当批次标准曲线6份lloq平均峰面积的20.0%,别嘌醇内标/氧别嘌醇内标干扰为0.0%~0.1%/0.0%;考察潜在药物干扰时,各浓度水平测定结果平均值的准确度在100
±
15%范围内,别嘌醇潜在药物干扰低/高浓度平均准确度为100.6%/98.5%,氧别嘌醇潜在药物干扰低/高浓度平均准确度为97.6%/101.2%,说明该分析方法
能准确区分别分析物别嘌醇和氧别嘌醇,别嘌醇对别嘌醇内标物无干扰,氧别嘌醇对氧别嘌醇内标物无干扰,别嘌醇和氧别嘌醇无潜在药物干扰。
[0228]
5.9人血浆样品检测
[0229]
于96孔板中加入300ul甲醇,加入50ul的is spike,精密加入50ul含别嘌醇及氧别嘌醇的血浆样品,涡旋混合3min,于4℃以2450g离心10min,2ul进样到色谱系统进行lc-ms/ms分析,结果如图 3~4所示。
[0230]
结论:经方法学验证,本发明方法具有灵敏度高,适用性强,精密度和准确度良好、稳定性好的优点,药物的提取回收率在可接受范围内,未见明显基质效应,无干扰,本发明方法还具有快速、通量高的优点。
[0231]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1