用于制备碘[的制作方法
用于制备碘[
131
i]化钠溶液的原料二氧化碲质量标准
技术领域
[0001]
本发明涉及一种质量标准,具体涉及用于制备碘[
131
i]化钠溶液的原料二氧化碲质量标准。
背景技术:
[0002]
二氧化碲是制备碘[
131
i]化钠溶液的主要原料,国内无药用标准,仅有《yst926高纯二氧化碲》行业标准,适用于制备电子元件,红外窗口、光纤放大器的高纯二氧化碲,标准中除外观质量外,二氧化碲的物理规格(粒径)和化学成分测试方法均无具体标准,注明供需双方协商确定标准,该标准不能满足作为药用原料的要求,通过在行业标准基础上结合药品注册时对关键原料的要求,拟定了二氧化碲的质量标准,能够科学合理的控制其质量,满足制备药品碘[
131
i]化钠溶液的要求。
技术实现要素:
[0003]
本发明所要解决的技术问题是现有二氧化碲的物理规格和化学成分测试方法均无具体标准,注明供需双方协商确定标准,该标准不能满足作为药用原料的要求,目的在于提供用于制备碘[
131
i]化钠溶液的原料二氧化碲质量标准,解决了现有二氧化碲的物理规格和化学成分测试方法均无具体标准,注明供需双方协商确定标准,该标准不能满足作为药用原料要求的问题。
[0004]
本发明通过下述技术方案实现:
[0005]
用于制备碘[
131
i]化钠溶液的原料二氧化碲质量标准,包括以下步骤:
[0006]
s1:根据二氧化碲为两性化合物的性质以及原子发射光谱中的te元素固有的特征谱线用于鉴别二氧化碲;
[0007]
s2:碲为矿物或矿渣中提取,导致原料中含有的元素杂质较多,从药品的安全性角度考虑,将有毒有害易引入碘[
131
i]化钠溶液中的元素杂质列入检测标准;
[0008]
s3:采用氧化还原滴定法测定原料中二氧化碲的含量。
[0009]
本发明的工作原理为:本技术按药品注册要求拟定关键原料的质量标准,使标准中各检测方法具体化,可执行;
[0010]
根据二氧化碲的理化性质制定鉴别项;
[0011]
根据二氧化碲的生产工艺分析杂质,制定检查项;
[0012]
明确含量测定方法,简便易行;
[0013]
制定符合药典模式、项目完善、限度合理的质量标准。
[0014]
本标准用于制备药品的二氧化碲的质量控制,有针对性地规定检测项目,切实加强对原料内在质量的控制,检测方法的适用性良好,既考虑了目前较关注的元素杂质,又采用了较为灵敏的仪器检测方法,体现了准确、灵敏、简便、快速的原则,通过二氧化碲两性化合物的性质以及原子发射光谱中的te元素固有的一系列特征谱线用于原料的鉴别,通过检查易引入碘[
131
i]化钠溶液中的元素杂质,进一步控制原料的质量,最后通过氧化还原滴定
法测定原料中二氧化碲的含量,形成严谨科学的质量控制体系,使得当原料方提供二氧化碲原料后,按照本技术标准便于检测提供的二氧化碲原料是否可用于药用原料。
[0015]
进一步地,原料中二氧化碲的含量为99.0%~101.0%。
[0016]
进一步地,通过溶解度法鉴别,二氧化碲符合两性化合物的性质。
[0017]
进一步地,通过icp法定性,二氧化碲在te特征谱线处有强度响应。
[0018]
进一步地,通过icp法定量,原料中元素杂质以及其它重金属的含量。
[0019]
进一步地,原料中元素杂质包括pb、cr、as、hg、cu、se。
[0020]
进一步地,原料中元素杂质铅含量小于等于0.5ppm、铬含量小于等于100ppm、砷含量小于等于1.5ppm、汞含量小于等于3ppm、铜含量小于等于100ppm、硒含量小于等于15ppm,其它有害元素总计含量小于等于20ppm。
[0021]
本发明与现有技术相比,具有如下的优点和有益效果:
[0022]
1、本发明用于制备碘[
131
i]化钠溶液的原料二氧化碲质量标准,为药用原料的生产提供质量可控、合格安全的原料;
[0023]
2、本发明用于制备碘[
131
i]化钠溶液的原料二氧化碲质量标准,本标准的各项目制定科学合理;
[0024]
3、本发明用于制备碘[
131
i]化钠溶液的原料二氧化碲质量标准,本标准检查项采用药典通则收载的检测方法,限度制订合理;
[0025]
4、本发明用于制备碘[
131
i]化钠溶液的原料二氧化碲质量标准,本标准含量测定方法简单易行,经验证准确度、精密度良好。
附图说明
[0026]
此处所说明的附图用来提供对本发明实施例的进一步理解,构成本技术的一部分,并不构成对本发明实施例的限定。在附图中:
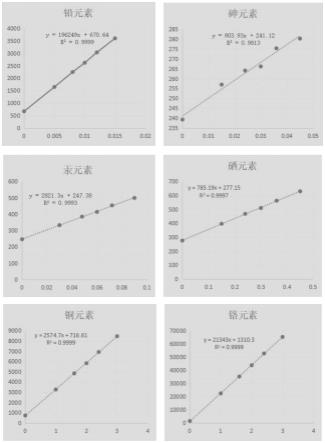
[0027]
图1为本发明元素杂质定量测定的线性图。
具体实施方式
[0028]
为使本发明的目的、技术方案和优点更加清楚明白,下面结合实施例和附图,对本发明作进一步的详细说明,本发明的示意性实施方式及其说明仅用于解释本发明,并不作为对本发明的限定。
[0029]
实施例1
[0030]
本发明用于制备碘[
131
i]化钠溶液的原料二氧化碲质量标准,本实施例的具体实现方式为:
[0031]
【性状】本品为白色粉末或结晶性粉末,无臭。
[0032]
【鉴别】(1)取本品约0.1g,加氢氧化钠试液适量使溶解,再滴加稀硫酸试液适量至显微酸性,有白色沉淀析出,继续滴加稀硫酸试液,沉淀溶解。
[0033]
(2)本品的原子发射光谱图中,应在214.2nm、225.9nm和238.5nm处有te元素的系列特征谱线。
[0034]
【检查】氯化物:取本品0.50g,依法检查(《中国药典》2020版四部通则0801),与标准氯化钠溶液2.5ml制成的对照液比较,不得更浓(0.005%)。
[0035]
水分:取本品,照水分测定法(《中国药典》2020版四部通则0832第二法)测定,含水分不得过0.5%。
[0036]
重金属及有害元素:照电感耦合等离子体原子发射光谱法(《中国药典》2020版四部通则0411)测定,铅含量小于等于0.5ppm、铬含量小于等于100ppm、砷含量小于等于1.5ppm、汞含量小于等于3ppm、铜含量小于等于100ppm、硒含量小于等于15ppm,其它有害元素总计含量小于等于20ppm。
[0037]
【含量测定】取本品约0.15g,精密称定,置250ml锥形瓶中,加5%氢氧化钠溶液10ml,加热使溶解至澄清,冷却后加入混合酸溶液10ml,摇匀,精密加入重铬酸钾滴定液(0.1mol/l) 25ml,摇匀,放置30min,用硫酸亚铁铵滴定液(0.1mol/l)滴定至淡黄色,加入二苯胺磺酸钠指示液5滴,继续滴定至溶液由紫色变为绿色即为终点,并将滴定的结果用空白校正。
[0038]
【贮藏】密封,在干燥处保存。
[0039]
实施例2
[0040]
实施例1的基础上,如图1所示,本实施例的具体实现方式为:
[0041]
概述
[0042]
已按照《元素杂质检测方法的建立和验证实验方案》建立了原料二氧化碲中重金属以及其它元素杂质的检测方法并进行验证。
[0043]
杂质谱分析
[0044]
高纯二氧化碲的行业标准中列出了化学成分,元素杂质有na、mg、al、ca、cr、mn、fe、cu、zn、ag、se、sn、pb、bi和ni等。
[0045]
二氧化碲中沸点较低的、易挥发的元素杂质及有害金属等,自然界广泛存在的砷盐和重金属,这些元素杂质均可能引入碘[
131
i]化钠制剂中,参考日本药典收载的碘[
131
i]化钠制剂中对元素杂质hg进行了控制,因此将pb、se、cu、cr、hg和as列入检测。
[0046]
杂质限度的确定
[0047]
待测元素杂质在ich q3d(r1)中的分类及口服pde(每日暴露量)值见表1。
[0048]
根据碘[
131
i]化钠溶液生产工艺,40g原料二氧化碲经辐照后可获得4ci的碘[
131
i]化钠溶液。假设所有的杂质无损失转移至终产品中,以200mci的最大日服用剂量计,相当于2g原料中含有的元素杂质应为pde,见表1。
[0049]
表1元素杂质pde及限度
[0050]
组分类别口服pde(μg/天)限度(μg/g)pb1类50.5as1类151.5hg1类303se2b类15015cu3类3000100cr3类11000100
[0051]
由上表可知,根据pde计算出cu、cr两种元素限度分别为1500μg/g和5500μg/g。为严格控制原料二氧化碲的质量,将cu、cr两种元素限度定为100μg/g。
[0052]
检测方法的建立
[0053]
消解条件的选择
[0054]
采用不同消解方式,对原料二氧化碲进行消解,对比不同消解方法之间的差异,选择能够使样品完全溶解的消解剂。结果见表2。
[0055]
表2不同消解剂的比较
[0056][0057]
由上述实验结果,选择浓盐酸为溶剂进行消解,确保完成检测前,溶液均匀,无沉淀,保证结果的准确性。
[0058]
元素杂质特征谱线的选择
[0059]
暂定辅气流速为0.5l/min,选择上述元素的3条灵敏线作为其特征谱线,依次分析空白样品、对照品溶液,比较两个样品在各波长下的谱线强度,选择灵敏度合适且干扰小的谱线作为其最佳特征谱线。筛选结果见表3。
[0060]
表3元素杂质特征谱线筛选结果
[0061][0062][0063]
各待测元素选择无其它元素干扰,且响应值明显高于空白的波长为特征谱线,根据实验结果,pb选择220.3nm,as选择193.7nm,hg选择194.2nm,se选择196.0nm,cu选择 224.7nm,cr选择267.7nm。
[0064]
辅助气体流量的选择
[0065]
分析软件参数设置中辅助气体流量可设置为0.5l/min、1.0l/min、1.5l/min,通过改变辅助气体流量,依次检测对照品溶液,比较不同辅助气体流量下的谱线强度,选择谱线强度响应值大的所对应的辅助气体流量作为检测方法的参数。结果见表4。
[0066]
表4不同辅气流量下元素杂质响应值
[0067][0068]
根据实验结果,综合考虑辅气流速对元素杂质谱线强度的影响,选择辅助气体流量为 0.5l/min。
[0069]
供试液浓度选择
[0070]
以限度控制最严格的pb元素为代表。
[0071]
由pb的检测限(3sd/k),计算供试品溶液浓度(溶液浓度=检测限/限度)。
[0072][0073]
即供试品溶液的浓度应不低于0.0001g/ml,在拟定的检测方法中,原料溶液的浓度为 0.02g/ml,满足测定要求,能够确保pb的检出。
[0074]
检测方法的验证
[0075]
各待测元素在原料中为痕量或微量,拟采用限度法测定,验证的指标有系统适用性、专属性、线性、检测限、定量限和溶液稳定性。
[0076]
待验证的检测方法
[0077]
经消解剂、特征谱线、辅助气体流量的筛选,确定元素杂质的检测方法如下:
[0078]
空白溶液:取浓盐酸5ml,用1%硝酸溶液稀释至50ml,摇匀。
[0079]
对照品溶液:精密量取铅标准溶液(100μg/ml)0.5ml、砷标准溶液(1000μg/ml)0.15ml、汞标准溶液(1000μg/ml)0.3ml和硒标准溶液(1000μg/ml)1.5ml,置于同一50ml量瓶中,用1%硝酸溶液稀释至刻度,摇匀,即得贮备液a。精密量取贮备液a 5.0ml、铜标准溶液 (100μg/ml)10ml、铬标准溶液(1000μg/ml)1ml,置于同一50ml容量瓶中,用1%硝酸溶液稀释至刻度,摇匀,得贮备液b。精密量取对照品贮备液b 5ml置于50ml容量瓶中,加入浓盐酸5ml,用1%硝酸溶液稀释至刻度,摇匀,即得。
[0080]
原料溶液:取本品1.0g,精密称定,加入浓盐酸5ml使溶解,用1%硝酸溶液稀释至50ml,摇匀。
[0081]
仪器参数:rf:1150w;泵速:45r/min;辅助气体流量:0.5l/min;雾化气体流量:开。
[0082]
测定法取空白溶液、对照品溶液及原料溶液,注入仪器,于元素杂质的特征分析谱线下测定,记录响应值。
[0083]
限度原料溶液如在元素特征分析谱线处有响应,其响应值不得大于对照品溶液中相应元素响应值。
[0084]
元素pbashgsecucr特征谱线/nm220.3193.7194.2196.0224.7267.7限度μg/g0.51.5315100100
[0085]
系统适用性
[0086]
在拟定条件下,取空白溶液、专属性溶液分别测定,记录数据。结果见表5。
[0087]
表5系统适用性结果
[0088][0089]
连续测定5次,元素杂质响应值的rsd为0.36~1.11%,符合要求(≤10%),系统适用性良好。
[0090]
专属性
[0091]
取空白溶液、原料溶液、对照品溶液及原料加标溶液,分别测定,记录数据。结果见表 6。
[0092]
表6专属性结果
[0093][0094]
由实验结果可知,对照品溶液与空白溶液相比,元素杂质特征分析谱线处有明显响应;原料加标溶液与原料溶液相比,有明显响应。表明空白溶液在元素杂质特征分析谱线处无干扰,元素杂质间也无干扰,该方法的专属性符合要求。
[0095]
线性
[0096]
取对照品贮备液2,分别精密量取0ml、2.5ml、4ml、5ml、6ml、7.5ml置于各50ml容量瓶中,加入浓盐酸5ml,用1%硝酸溶液稀释至刻度,摇匀,即得系列线性溶液,按拟定的条件试验,分别对空白溶液、系列对照品溶液进行测定,结果见表7,图1。
[0097]
表7元素杂质线性数据
[0098][0099]
定量限与检测限
[0100]
制备空白溶液10份,按拟定的条件试验,对空白溶液进行测定,计算空白溶液响应值的标准偏差sd,结合各待测元素的线性斜率,计算元素杂质的定量限(10sd/k)和检测限(3sd/k)。
[0101]
定量限溶液:精密量取各单元素标准溶液适量,加入浓盐酸5ml,用1%硝酸溶液定量稀释制成含元素杂质为10sd/k(μg/ml)的溶液。
[0102]
检测限溶液:精密量取各单元素标准溶液适量,加入浓盐酸5ml,用1%硝酸溶液定量稀释制成含元素杂质为3sd/k(μg/ml)的溶液。
[0103]
分别将10份空白溶液、定量限溶液和检测限溶液注入仪器,记录数据,计算结果见表 8~10。
[0104]
表8定量限与检测限计算结果
[0105][0106]
表9定量限验证
[0107][0108][0109]
表10检测限验证
[0110][0111]
由实验结果可知,当原料溶液浓度为0.02g/ml时,能够检出0.0025ppm的pb、0.195ppm 的as、0.08ppm的hg、0.3ppm的se、0.21ppm的cu和0.065ppm的cr;能够对0.009ppm 的pb、0.655ppm的as、0.275ppm的hg、0.995ppm的se、0.695ppm的cu和0.21ppm的cr 准确定量。
[0112]
元素杂质的定量限浓度均小于其限度浓度;元素杂质检测限浓度均小于限度浓度的0.5 倍,该方法能够满足测定要求。
[0113]
溶液稳定性
[0114]
将空白溶液、原料溶液和对照品溶液在室温下放置,于0h、0.5h、1h、1.5h、2h、4h分别取样,注入仪器,记录响应值。结果见表11。
[0115]
表11溶液稳定性数据
[0116][0117][0118]
0~2.5h室温下,以0h为基准,对照品溶液与对照品溶液其它各时间点待测元素的响应值均在70~130%范围内,且rsd≤10%,因此对照品溶液和原料溶液在室温下2.5h内稳定,2.5h 后原料溶液中有固体析出。
[0119]
验证总结
[0120]
综上所述,该检测方法的专属性、线性、检测限、定量限、溶液稳定性以及系统适用性的验证结果均符合要求,该检测方法可用于二氧化碲中的元素杂质的测定,汇总结果见表12。
[0121]
表12二氧化碲中元素杂质检测方法验证结果
[0122][0123]
综上所述,该检测方法的专属性、线性、检测限、定量限、溶液稳定性以及系统适用性的验证结果均符合要求,该检测方法可用于二氧化碲中的元素杂质的测定。
[0124]
样品测定
[0125]
按拟定方法对六批原料中的元素杂质进行检测,各元素响应值若低于对照品的响应值,即各元素含量均未超过限度,合格;反之则超限,不合格。结果见表13~15。
[0126]
表13原料中元素杂质测定结果
[0127][0128]
表14原料中元素杂质测定结果
[0129][0130]
表15原料中元素杂质测定结果
[0131][0132]
以上所述的具体实施方式,对本发明的目的、技术方案和有益效果进行了进一步详细说明,所应理解的是,以上所述仅为本发明的具体实施方式而已,并不用于限定本发明的保护范围,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1