吸头式微萃取注射液、口服液中糠醛类化合物的检测方法
1.本发明属于药物检测分析领域,涉及一种吸头式微萃取注射液、口服液中糠醛类化合物的检测方法。
背景技术:
2.5-羟甲基糠醛(5-hmf)、糠醛(f)、5-甲基呋喃-2-醛(mf)是含糖样品在储存过程中形成的化合物,常被认为是由于过度受热或储存条件的不佳而导致质量恶化的标志。在药品的生产和储存的过程中,也可能会生成糠醛类物质,如葡萄糖注射液在灭菌和储存期间主要热分解产物为5-hmf。有研究报道发现摄入超过规定剂量的5-hmf对人类健康构成潜在风险,如5-hmf能够与氨基酸形成加合物而产生细胞毒性。此外,5-hmf及其低聚物对动物和人体存在潜在的神经毒性,尤其在婴幼儿神经发育过程中。世界卫生组织和欧盟食品法典委员会将蜂蜜中的5-hmf的最高限量设定为40mg/kg,以确保产品在加工过程中未经加热变质。目前,有越来越多研究表明摄入过多5-hmf可能会对健康造成危害。因此,有必要建立适当的方法来测定含糖药品如注射液和口服液和食品中5-hmf的含量。
3.目前,检测糠醛类物质的方法有许多,de andrade jucimara等通过高效液相色谱法(hplc)检测到饼干、面包等含糖食品的5-hmf,jiaqi shi等采用了液相色谱与质谱联用法(lc-ms)检测到奶制品中的5-hmf。viviane maria rizelio等人通过胶束电动毛细管色谱(mekc)方法进行了蜂蜜中5-hmf测定。尽管使用hplc能直接检测糠醛类物质,但在其284nm紫外吸收处,易有其他杂质峰的干扰从而影响定量准确性。
4.影响检测糠醛类物质精确性的原因还包括检测实际样品中共存的基质较复杂,复杂的基质会干扰目标分析物的测定,因此使用有效的前处理技术提取目标分析物是非常有必要的。近年来,在复杂基质中提取5-hmf的分析方法层出不穷,主要包括液相萃取、固相萃取、液相微萃取、固相支撑液相萃取、薄膜微萃取等。传统的液相萃取法费时费力,萃取过程需要消耗大量的有机溶剂。然而,均相液-液萃取(hlle)以其操作简单、快速、有机溶剂消耗低等的优点,已成为最常用的样品制备方法之一。wenbin chen等人建立了盐析辅助液-液萃取法用于测定蜂蜜样品中的5-hmf,但该过程相对繁琐,需要一系列步骤,如混合旋涡和高速离心。固相萃取(spe)也常用于萃取和浓缩。但传统的固相萃取不能实现高效萃取,并且操作较为繁琐、成本较高。
技术实现要素:
5.为了克服现有技术的不足,本发明的目的在于提供吸头式微萃取注射液、口服液中糠醛类化合物的检测方法,该检测方法具有步骤简单,检测精度高的特点。
6.本发明的目的采用如下技术方案实现:
7.一种吸头式微萃取注射液、口服液中糠醛类化合物的检测方法,包括以下步骤:
8.(1)将内部纤维填料装入吸头式萃取装置内,然后将溶解有衍生化试剂的萃取溶剂装载于所述内部填料上;
9.(2)将注射液或口服液装载于步骤(1)的内部纤维填料上,再向所述萃取装置内添加解吸溶剂,得到含有待分析物衍生物的解吸液;
10.(3)将步骤(2)得到的含有待分析物衍生物的解吸液进行检测、分析。
11.进一步地,所述注射液为中药注射液或含糖注射液,所述口服液为含糖口服液。
12.进一步地,所述内部填料为木棉纤维,所述木棉纤维与所述注射液或口服液的添加比例为1-5mg:1ml。
13.进一步地,所述步骤(1)中衍生化试剂为2,4-二硝基苯肼,所述萃取溶剂选自乙酸乙酯、甲苯、二氯甲烷、三氯甲烷的一种或多种。
14.进一步地,所述乙酸乙酯与甲苯的体积比为0.25-3:1。
15.进一步地,所述衍生化试剂与萃取溶剂的添加比例为0.1-10mg:1ml。
16.进一步地,步骤(2)装载过程为将注射液或口服液多次吸打在所述内部纤维填料上;
17.所述添加解吸溶剂的过程为将解吸溶剂多次吸打在所述内部填料上。
18.进一步地,所述步骤(3)的解吸溶剂为甲醇、乙腈、乙醇、丙酮中的一种。
19.进一步地,所述糠醛类化合物为5-羟甲基糠醛、5-甲基-2-糠醛或糠醛。
20.相比现有技术,本发明的有益效果在于:
21.本发明提供了一种吸头式微萃取注射液、口服液中糠醛类化合物的检测方法,该方法将溶解有衍生化试剂的萃取溶剂添加至内部纤维填料上,然后以多次吸打的将注射液或口服液装载于步骤(1)的内部纤维填料上,使衍生化和萃取过程同步进行。然后向内部填料上以多次吸打的方式添加解吸溶剂,最终将得到含有待分析物衍生物的解吸液进行检测、分析。该方法具有较好的萃取效果,回收率稳定,能够满足一定条件下的快速处理分析,具有较强的开发潜力。和其他分析检测方法相比,该方法的灵敏度和选择性好,实验操作简单,成本低廉,可实现快速、自动化的萃取分析。
附图说明
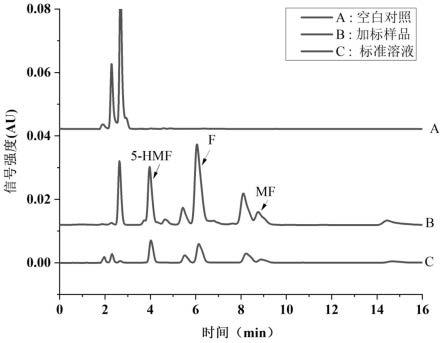
22.图1为本发明实施例1、对比例1-2的可行性分析hplc-uv色谱图;
23.图2为本发明实施例2不同萃取剂种类对应的检测结果图;
24.图3为本发明实施例2萃取剂中乙酸乙酯与甲苯的不同体积比对应的检测结果图;
25.图4为本发明实施例3木棉纤维的不同添加量对应的检测结果图;
26.图5为本发明实施例4衍生化试剂的不同添加量对应的检测结果图;
27.图6为本发明实施例5甲酸的不同含量对应的检测结果图;
28.图7为本发明实施例6不同解吸溶剂对应的检测结果图;
29.图8为本发明实施例7解吸溶剂的不同添加量对应的检测结果图;
30.图9为本发明实施例8上样吸打次数对应的检测结果图;
31.图10为本发明实施例9解吸吸打次数对应的检测结果图;
32.图11为本发明实施例检测过程示意图。
具体实施方式
33.下面,结合附图以及具体实施方式,对本发明做进一步描述,需要说明的是,在不
相冲突的前提下,以下描述的各实施例之间或各技术特征之间可以任意组合形成新的实施例。
34.实施例1
35.吸头式微萃取检测糠醛类化合物模型的建立,包括以下步骤:
36.(1):
37.(1.1)吸头式微萃取装置的制备:萃取装置是由200μl移液器吸头和1000μl移液器吸头组装而成,准确称取3.00mg木棉纤维于200μl移液吸头中,将1000μl移液器吸头尖端在适当的位置切断,并将其与200μl移液器吸头的顶部连接在一起,即制备成吸头式微萃取装置。木棉纤维将被卡在组合吸头的中下部(连接处以下),从而避免后续样品溶液和解吸液吸打过程中木棉纤维随溶液而上下移动。
38.(1.2)分析物标准溶液的配制:用十万分之一天平精密称取5-hmf(5-羟甲基糠醛)、f(糠醛)、mf(5-甲基-2-糠醛)适量,分别置于不同ep管中,加入分析纯乙腈,涡旋混匀,配制成5mg/ml质量浓度的储备液,转移至安瓿瓶中备用;分别吸取上述3种醛类储备液适量,加入一定量超纯水,并添加体积比0.1%的甲酸,配制成10μg/ml的混合标准品溶液(含0.1%甲酸),放入4℃冰箱备用。
39.(1.3)样品溶液的预处理:注射液、口服液等样品中直接添加体积比0.1%的甲酸备用。
40.(1.4)衍生化试剂溶液的制备:本发明微萃取和衍生化同时进行,根据2,4-二硝基苯肼(dnph)的溶解性,选择乙酸乙酯(ea)作为萃取溶剂。精确称取dnph适量,加入分析纯乙酸乙酯,涡旋混匀,配制成1mg/ml质量浓度的衍生化试剂溶液。由于乙酸乙酯具有挥发性,衍生化试剂浓度易发生改变,故用封口膜封口4℃冰箱保存。
41.(2)吸头式微萃取:准确吸取步骤(1.4)得到的12.5μl衍生化试剂溶液装载于内部纤维填料
‑‑
木棉纤维上,采用移液器装配制备好的萃取装置,上样过程通过按压/松开移液器压杆,吸打1.0ml混合标准品溶液(含0.1%甲酸)或者样品溶液(含有0.1%甲酸),反复缓慢吸打20次,此过程同时完成衍生化和萃取,最后用移液器准确吸取200μl甲醇(meoh)进行解吸,反复解吸吸打15次,将所得含有待分析物衍生物的解吸液直接进行hplc-uv检测。
42.(3)检测:采用agilent 5tc-c18色谱柱,型号为4.6mm
×
150mm,5μm,流动相a:超纯水,流动相b:色谱级甲醇,等梯度洗脱,a:b=30:70(v/v),柱温控制在25℃,进样量设定10μl,流速设定为1.0ml/min,紫外检测器波长设定为360nm。
43.经多次试验验证,待分析物衍生物与待分析物的摩尔比为0.9-1:1。
44.为了验证实验方案的可行性,每个实验均重复3次,下同。
45.对比例1
46.对比例1与实施例1的区别在于:对比例1为空白衍生萃取,具体是将实施例1中的样品溶液替换为纯水(含0.1%甲酸)。
47.对比例2
48.对比例2与实施例1的区别在于:对比例2为直接衍生化实验,即该分析物标准溶液直接和衍生化溶液混合,不经过本发明的萃取装置进行萃取,将得到的衍生物溶液直接分析检测。具体为省略实施例1中步骤(1)、(2),向1.0ml步骤(2)得到的分析物标准溶液中直接加入12.5μl步骤(1.2)得到的衍生化试剂溶液,混合涡旋30s,室温放置30min。
49.为了考察吸头式萃取装置性能,分别对分析物标准溶液分析物经过萃取装置萃取(实施例1)、空白衍生萃取(对比例1)、不经过萃取装置萃取(对比例2)进行了考察,如图1所示,图1为实施例1、对比例1-2的可行性分析hplc-uv色谱图。结果表明,本发明的检测方法能检测到所有目标分析物,并且空白对照没有出现干扰分析物的峰,同时做了三组平行实验考察重复性,rsd在10%之内,因此,本研究提供的萃取装置能有效用于样品前处理。
50.实施例2
51.本实施例与实施例1的区别在于:将步骤(1.2)中的萃取溶剂乙酸乙酯分别调整为甲苯(eb)、二氯甲烷(mb)、三氯甲烷(tcm),其余步骤与实施例1相同。
52.检测结果如图2所示,乙酸乙酯和甲苯萃取5-hmf效果相当,甲苯萃取f和mf最好,可能原因是乙酸乙酯有弱极性,而f和mf衍生化后非极性的性质比5-hmf大,因此f和mf在非极性大的甲苯中溶解溶解度高,用甲苯萃取效果较好。
53.根据上述结果,将萃取溶剂乙酸乙酯调整为乙酸乙酯和甲苯的混合溶剂,其中乙酸乙酯与甲苯的体积比分别为75:25、50:50、20:80。其余步骤与实施例1相同。
54.检测结果如图3所示,结果表明采用乙酸乙酯与甲苯的混合溶剂做萃取溶剂,其萃取效果相比使用单一萃取溶剂的效果有一定程度的提升。
55.实施例3
56.本实施例与实施例1的区别在于:将实施例1步骤(1.1)木棉纤维用量分别调整为1mg、2mg、2.5mg、4mg、5mg,衍生化试剂的添加量为12.5μl,标准品溶液为1ml,其余步骤与实施例1相同。
57.检测结果如图4所示,当衍生化试剂的添加量为12.5μl,标准品溶液为1ml时,木棉纤维的用量大于2mg时,有助于增加待分析物的回收率。
58.实施例4
59.本实施例与实施例1的区别在于:将实施例1步骤(2)的衍生化试剂的用量分别调整为5μl、7.5μl、10μl、15μl、20μl、25μl,其余与实施例1相同。
60.检测结果如图5所示,当衍生化试剂的用量增加时,5-hmf衍生化产物逐渐增加,回收率也随着增加。当衍生化试剂的用量为15μl时,萃取效率达到最高,然而,当衍生化试剂的用量继续增加时,萃取回收率反而急剧下降。可能是由于木棉纤维的量是一定的,随着衍生化试剂量的增加,过量的衍生化试剂在吸打衍生/萃取过程中会从木棉纤维上掉落,导致萃取效果变差。
61.实施例5
62.本实施例与实施例1的区别在于:将实施例1步骤(1.1)中分析物标准溶液中甲酸的体积含量分别调整为0.5%、1.0%、2.0%、3.0%、5.0%,其余与实施例1相同。
63.检测结果如图6所示,甲酸含量增加至1%时,生成的衍生产物峰面积达到最大值,萃取回收率最高。这是因为醛类化合物和dnph反应生成的腙类物质,是在偏酸性条件下进行的,对酸性环境有一定的要求,实验结果表明,用甲酸调节反应体系的ph时,当添加量为1%时,最有利于萃取过程的进行。
64.实施例6
65.解吸溶剂种类及用量的优化
66.本实施例与实施例1的区别在于:将实施例1中的解吸液调整为甲醇(meoh)、乙醇
(etoh)、乙腈(acn)、丙酮(ac)。
67.目标分析物是具有极性的,被衍生化后具有了疏水性质,因此解吸溶剂必须满足对目标分析物衍生化产物的的良好溶解度。检测结果如图7所示,当以乙腈和丙酮为解吸溶剂时,萃取回收率较好。
68.实施例7
69.本实施例与实施例1的区别在于:将实施例1步骤(2)中的甲醇用量分别调整为100μl、150μl、250μl、300μl,其余与实施例1相同。
70.检测结果如图8所示,当解吸液用量增加时,5-hmf衍生产物的量逐渐增加,回收率也在逐渐增大,当解吸液用量为200μl时,萃取效率基本达到恒定。解吸液用量过少,目标分析物解吸不完全,萃取效率较低。本发明的解吸液用量符合微萃取使用较少量解吸液即可实现较大的富集因子实现最大程度萃取的要求。
71.实施例8
72.本实施例与实施例1的区别在于:将实施例1步骤(2)中上样吸打次数分别调整为5次、10次、15次、30次,其余步骤与实施例1相同。
73.检测结果如图9所示,当上样吸打次数增加时,萃取回收率逐渐增大,上样吸打20次时,回收率达到最高,萃取效率基本达到恒定。当吸打系数较少时,衍生化试剂和萃取剂不能完成与分析物的充分的接触,导致目标分析物的衍生化及萃取不完全,萃取回收率较低。
74.实施例9
75.本实施例与实施例1的区别在于:将实施例1步骤(2)中解吸吸打次数分别调整为5次、10次、15次、30次,其余步骤与实施例1相同。
76.检测结果如图10所示,随着解吸吸打次数的增加,衍生产物的量随之增加,萃取回收率不断的增大,但增高趋势并不明显,当解吸吸打15次时,分析物已基本完全解吸下来。
77.实验例1
78.1.1本发明检测方法的线性关系和检测限
79.5-hmf用水配成0.5μg/ml、1μg/ml、2μg/ml、5μg/ml、10μg/ml、20μg/ml的一系列溶液,f用水配成0.1μg/ml、0.2μg/ml、1μg/ml、2μg/ml、5μg/ml、10μg/ml、20μg/ml的一系列溶液,mf用水配成1μg/ml、2μg/ml、5μg/ml、10μg/ml、20μg/ml的一系列溶液,按照实施例1中步骤(2)、步骤(3)的方法进行分析,记录峰面积(每个浓度3组平行最后取平均值),最后以峰面积均值对分析物浓度线性回归,以信噪比3:1和10:1分别计算检测限(lod)和定量限(loq),如表1所示,结果表明5-hmf、f、mf分别在0.5~20μg/ml、0.1~20μg/ml、1~20μg/ml浓度范围内线性关系良好,r2分别为0.990、0.996、0.993。
80.表1
81.[0082][0083]
1.2本发明检测方法的精密度和准确度
[0084]
5-hmf、f、mf用水分别配制成1μg/ml、5μg/ml、10μg/ml低中高三个质量浓度按照实施例1中步骤(2)、步骤(3)的方法进行分析(每个浓度3组平行),纪录峰面积,根据峰面积计算精密度,将峰面积带入各自标准曲线计算准确度,如表4,结果显示准确度分别均在84%~116%范围内,rsd分别均在10%以内。
[0085]
表2
[0086][0087]
实验例2
[0088]
吸头式微萃取注射液和口服液中糠醛类化合物的检测方法
[0089]
注射液和口服液中糠醛类化合物的检测方法过程如下:取实际注射液或口服液样品1ml,加入1μl甲酸,混匀后得到1ml含有0.1%甲酸的样品溶液,然后再按照实施例1中步骤(2)、(3)处理和检测,检测示意图如图11所示。
[0090]
结果如表3所示,5%葡萄糖注射液、清开灵注射液、双黄连注射液、葡萄糖酸锌口服液中均检测到了5-hmf,而另外两种糠醛(f和mf)均未检测到。
[0091]
表3
[0092]
样品5-hmf(μg/ml)f(μg/ml)mf(μg/ml)5%葡萄糖注射液0.33n.d.n.d.清开灵注射液0.01n.d.n.d.双黄连注射液9.21n.d.n.d.葡萄糖酸锌口服溶液3.77n.d.n.d.
[0093]
在一些应用场景中,本发明的检测方法还可以用于检测食品中糠醛类物质,如苹果汁、混合果蔬饮料、蜂蜜。
[0094]
其中样品的制备过程如下:
[0095]
苹果汁:准确称取苹果汁试样500μl,加入1μl甲酸,混匀后得到1ml含有0.1%甲酸的样品溶液。
[0096]
50%混合果蔬:苹果汁试样500μl,加入1μl甲酸,混匀后得到1ml含有0.1%甲酸的
样品溶液。
[0097]
蜂蜜样品:准确称取蜂蜜样品0.1g(精确至0.01g),移液枪吸取超纯水将蜂蜜稀释至1ml,再加入,加入1μl甲酸,混匀后得到1ml含有0.1%甲酸的样品溶液。
[0098]
检测过程参照实施例1步骤(2)、步骤(3)。
[0099]
结果如表4所示,在蜂蜜中检测到5-hmf,含量为0.86μg/ml,根据蜂蜜样品制备200μg/ml浓度计算相当于0.43mg/kg,未超过国际标准蜂蜜中的5-hmf的最高限量40mg/kg。在苹果汁、蜂蜜中均检测到f,表明该方法可用于实际含糖溶液样品中5-hmf、f的含量测定。
[0100]
表4
[0101]
样品5-hmf(μg/ml)f(μg/ml)mf(μg/ml)苹果汁n.d.0.97n.d.50%混合果蔬饮料n.d.n.d.n.d.蜂蜜0.860.99n.d.
[0102]
上述实施方式仅为本发明的优选实施方式,不能以此来限定本发明保护的范围,本领域的技术人员在本发明的基础上所做的任何非实质性的变化及替换均属于本发明所要求保护的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1