一种药物中残留小分子有机物的检测方法与流程
1.本技术属于药物分析技术领域,尤其涉及一种药物中残留小分子有机物的检测方法。
背景技术:
2.药品的残留溶剂,又称有机挥发性杂质,是指在活性药物成分、辅料和药品生产过程中使用和产生的有机挥发性化学物质。药品还可被来自包装、运输、仓储中的有机溶剂污染。药品生产商有责任确保终产品中的残留溶剂含量在符合规定的限度内。根据gb/t 24395-2009食品工业用吸附树脂标准可确定氯苯、甲基丙烯酸甲酯、甲苯、二甲苯、二乙烯苯、苯乙烯、苯、1,2-二氯乙烷的限度浓度,根据中国药典2020年版四部0861残留溶剂测定法通则可确定正庚烷、乙醇的限度浓度。
3.目前,药物中残留溶剂的检测方法多样,但大多是针对一类残留溶剂制定的检测方案,对于存在多种残留溶剂污染的药物体系需要大量重复试验,检测方案缺乏普适性。同时,针对一些小分子有机溶剂的检测方案灵敏度较差,难以准确评估药物中残留溶剂的安全性。
技术实现要素:
4.有鉴于此,本技术提供了一种药物中残留小分子有机物的检测方法,建立了药物中10种小分子残留有机物的含量测定方案,解决了现有药物中残留溶剂检测方法普适性低、灵敏度差的问题。
5.本技术的具体技术方案如下:本技术提供一种药物中残留小分子有机物的检测方法,包括如下步骤:设定气相色谱和质谱条件参数,其中气相色谱条件参数中气相色谱柱为中极性气相色谱柱,进样方式为顶空进样,质谱条件参数中扫描方式为sim扫描;配制药物供试品溶液和对照品溶液,其中采用混合有机溶液处理药物供试品和对照品,混合有机溶液为二甲基亚砜和甲醇的混合溶液;将药物供试品溶液和对照品溶液在气相色谱-质谱条件下进样检测,并分析药物供试品中残留小分子有机物的含量。
6.进一步的,所述气相色谱条件参数中柱温升温程序为:起始温度40~50℃,保持10~15min,10℃/min升至100~150℃,20℃/min升至200~250℃,保持1~2min,20℃/min升至250~280℃,保持2~5min。
7.进一步的,所述气相色谱条件参数中分流模式为:分流比10~50:1。
8.进一步的,所述质谱条件参数中扫描参数如下:采集组1:离子m/z为29、31、45、46,采集组2:离子m/z为26、52、53,采集组3:离子m/z为27、41、43、49、51、57、62、64、71、77、78,采集组4:离子m/z为39、41、63、65、69、91、92、100,采集组5:离子m/z为51、77、78、91、103、104、105、106、112、114,采集组6:离子m/z为
624ms,60m
×
0.25mm
×
1.40μm;分流模式:分流比20:1;进样方式:顶空进样;柱温:起始温度45℃,保持10min,10℃/min升至150℃,20℃/min升至200℃,保持2min,20℃/min升至250℃,保持2.5min。
22.2、质谱条件参数:离子源:ei;离子源温度:230℃;四级杆温度:150℃;传输线温度:260℃;扫描方式:sim;扫描参数如下:采集组1:离子m/z为29、31、45、46,采集组2:离子m/z为26、52、53,采集组3:离子m/z为27、41、43、49、51、57、62、64、71、77、78,采集组4:离子m/z为39、41、63、65、69、91、92、100,采集组5:离子m/z为51、77、78、91、103、104、105、106、112、114,采集组6:离子m/z为115、128、129、130。
23.3、溶液配制:空白溶液:量取100ml二甲基亚砜加入到500ml烧杯中,再量取100ml甲醇加入,混匀,移取2ml至20ml顶空瓶,压盖即得体积比为1:1的二甲基亚砜-甲醇溶液。
24.供试品溶液:精密称取红霉素样品约0.2g,置于20ml顶空瓶中,加入2ml空白溶液使完全溶解,压盖,摇匀即得。
25.检测限溶液:分别精密称取正庚烷、乙醇、氯苯、甲基丙烯酸甲酯、甲苯、二甲苯、二乙烯苯、苯乙烯、苯、1,2-二氯乙烷对照品约80mg,加少量二甲基亚砜溶解,再用空白溶液稀释,摇匀即得混合对照品储备液。精密移取0.2ml混合对照品储备液于10ml容量瓶中,用空白溶液稀释至刻度,摇匀即得。各个对照品配制的检测限溶液浓度见表1所示。
26.表1
标准曲线溶液:分别移取混合对照品储备液适量于不同容量瓶中,用空白溶液稀释至刻度,摇匀,即得一系列不同浓度标准曲线溶液。各个对照品配制的标准曲线溶液浓度见表2所示。
27.表2100%限度浓度加标供试品溶液:精密称取红霉素样品约0.2g,置于干净顶空瓶中,准确加入2ml标准曲线溶液(表2)中std3浓度对应溶液,使溶解,压盖,摇匀即得。
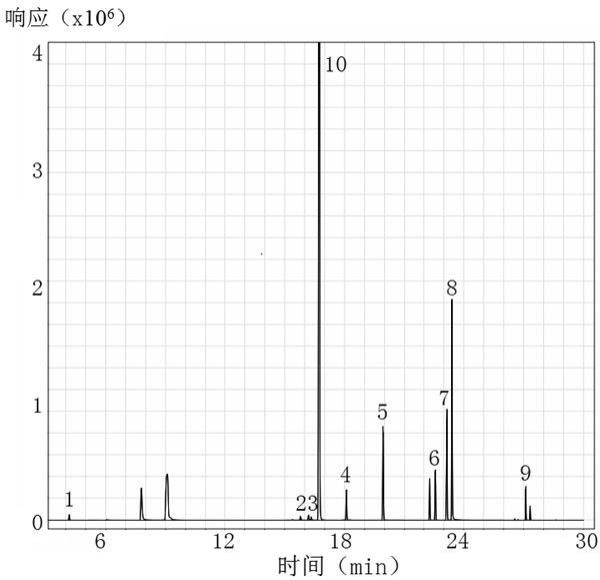
28.二、方法结果:1、专属性:参照上述方法条件,分别取空白溶液、供试品溶液、std3浓度对照品溶液以及100%限度浓度加标供试品溶液进样检测,记录色谱图。加标供试品溶液检测结果见图1所示。结果表明,空白溶液色谱图中未检出目标峰,对检测无干扰;供试品溶液色谱图中未检出目标峰;100%限度浓度对照品溶液和100%限度浓度加标供试品溶液色谱图中显示目标峰;结果符合规定,该方法专属性良好。同时,10种小分子待测物分离度良好,能够用于红霉素样品中10种小分子物质的同步含量测定。
29.2、线性:参照上述方法条件,分别取各个浓度下的标准曲线溶液进样检测。结果表明:正庚烷在15.2286μg/ml~101.5240μg/ml范围内,约相当于限度浓度的30%~200%,正庚烷峰面积与浓度呈良好的线性,相关系数r为0.9930;乙醇在14.7769μg/ml~98.5130μg/ml范围内,约相当于限度浓度的30%~200%,乙醇峰面积与浓度呈良好的线性,相关系数r为0.9994;氯苯在0.3139μg/ml~2.0926μg/ml范围内,约相当于限度浓度的30%~200%,氯苯峰面积与浓度呈良好的线性,相关系数r为0.9997;甲基丙烯酸甲酯在0.6148μg/ml~4.0986μg/ml范围内,约相当于限度浓度的30%~200%,甲基丙烯酸甲酯峰面积与浓度呈良好的线性,相关系数r为0.9999;甲苯在0.6181μg/ml~4.1208μg/ml范围内,约相当于限度浓度的30%~
200%,甲苯峰面积与浓度呈良好的线性,相关系数r为0.9998;二乙烯苯在0.3136μg/ml~2.0909μg/ml范围内,约相当于限度浓度的30%~200%,二乙烯苯峰面积与浓度呈良好的线性,相关系数r为0.9992;二甲苯在0.5213μg/ml~3.4752μg/ml范围内,约相当于限度浓度的30%~200%,二甲苯峰面积与浓度呈良好的线性,相关系数r为0.9999;苯乙烯在0.6542μg/ml~4.3613μg/ml范围内,约相当于限度浓度的30%~200%,苯乙烯峰面积与浓度呈良好的线性,相关系数r为1.0000;苯在0.0516μg/ml~0.3442μg/ml范围内,约相当于限度浓度的30%~200%,苯峰面积与浓度呈良好的线性,相关系数r为0.9997;1,2-二氯乙烷在0.0548μg/ml~0.3654μg/ml范围内,约相当于限度浓度的30%~200%,1,2-二氯乙烷峰面积与浓度呈良好的线性,相关系数r为0.9993;线性结果符合规定。
30.3、检测限:参照上述方法条件,取检测限溶液连续进样3次,记录色谱图。结果表明:连续3针检测限溶液中正庚烷浓度为10.1524μg/ml,约相当于限度浓度的20%,s/n大于3;乙醇浓度为9.8513μg/ml,约相当于限度浓度的20%,s/n大于3;氯苯浓度为0.2093μg/ml,约相当于限度浓度的20%,s/n大于3;邻二甲苯浓度为0.1166μg/ml,约相当于限度浓度的20%,s/n大于3;甲基丙烯酸甲酯浓度为0.4099μg/ml,约相当于限度浓度的20%,s/n大于3;甲苯浓度为0.4121μg/ml,约相当于限度浓度的20%,s/n大于3;对二乙烯苯浓度为0.10455μg/ml,约相当于限度浓度的20%,s/n大于3;对/间二甲苯浓度为0.2310μg/ml,约相当于限度浓度的20%,s/n大于3;苯乙烯浓度为0.4361μg/ml,约相当于限度浓度的20%,s/n大于3;苯浓度为0.0344μg/ml,约相当于限度浓度的20%,s/n大于3;1,3-二乙烯苯浓度为0.10455μg/ml,约相当于限度浓度的20%,s/n大于3;1,2-二氯乙烷浓度为0.0365μg/ml,约相当于限度浓度的20%,s/n大于3。
31.参照上述方法条件,取std1浓度对照品溶液连续进样6次,记录色谱图。结果表明:连续6针定量限溶液中正庚烷浓度为15.2286μg/ml,约相当于限度浓度的30%,s/n大于10,峰面积的rsd(n=6)为0.74%;乙醇浓度为14.7769μg/ml,约相当于限度浓度的30%,s/n大于10,峰面积的rsd(n=6)为3.22%;氯苯浓度为0.3139μg/ml,约相当于限度浓度的30%,s/n大于10,峰面积的rsd(n=6)为3.49%,甲基丙烯酸甲酯浓度为0.6148μg/ml,约相当于限度浓度的30%,s/n大于10,峰面积的rsd(n=6)为1.49%,甲苯浓度为0.6181μg/ml,约相当于限度浓度的30%,s/n大于10,峰面积的rsd(n=6)为1.67%,二甲苯浓度为0.5213μg/ml,约相当于限度浓度的30%,其中邻二甲苯、间二甲苯、对二甲苯的s/n均大于10,峰面积的rsd(n=6)为2.91%;二乙烯苯浓度为0.3136μg/ml,约相当于限度浓度的30%,其中1,3-二乙烯苯、对二乙烯苯的s/n均大于10,峰面积的rsd(n=6)为9.50%;苯乙烯浓度为0.6542μg/ml,约相当于限度浓度的30%,s/n大于10,峰面积的rsd(n=6)为4.13%;苯浓度为0.0516μg/ml,约相当于限度浓度的30%,s/n大于10,峰面积的rsd(n=6)为1.65%;1,2-二氯乙烷浓度为0.0548μg/ml,约相当于限度浓度的30%,s/n大于10,峰面积的rsd(n=6)为1.88%;该方法检测限和定量限结果符合规定。
32.4、准确度:参照上述方法条件,取100%限度浓度加标供试品溶液分别进样6次进行分析,记录色谱图并计算回收率结果。结果表明: 100%限度浓度的6份加标供试品溶液中目标正庚烷的回收率范围为84.8%-92.1%,回收率的rsd(n=6)为3.33%;乙醇回收率范围为81.0%~91.1%,回收率的rsd(n=6)为4.27%;氯苯回收率范围72.9%~78.2%,回收率的rsd(n=6)为2.77%;甲酯丙烯酸甲酯简称的回收率范围为77.3%~82.9%,回收率的rsd(n=6)为
2.68%;甲苯的回收率范围为76.7%~81.2%,回收率的rsd(n=6)为76.74%;二甲苯的回收率范围为72.4%~79.3%,回收率的rsd(n=6)为3.64%;二乙烯苯的回收率范围为73.6%~81.8%,回收率的rsd(n=6)为4.33%;苯乙烯的回收率范围为73.3%~81.3%,回收率的rsd(n=6)为4.31%;苯的回收率范围为74.4%~79.7%,回收率的rsd(n=6)为2.45%;1,2-二氯乙烷的回收率范围为75.4%~80.1%,回收率的rsd(n=6)为2.50%;准确度结果符合规定。
33.5、稳定性:参照上述方法条件,分别取std1浓度对照品溶液、供试品溶液和100%限度浓度加标供试品溶液于室温条件下放置不同时间后进样分析,记录色谱图。结果表明,于室温条件放置28.5h,供试品溶液中均未检出正庚烷、乙醇、氯苯、甲基丙烯酸甲酯、甲苯、二甲苯、二乙烯苯、苯乙烯、苯、1,2-二氯乙烷;100%限度浓度对照品溶液中正庚烷的检测浓度与初始(0h)检测浓度的比值介于75.2%~90.2%之间,乙醇的检测浓度与初始(0h)检测浓度的比值介于75.1%~92.3%之间,氯苯的检测浓度与初始(0h)检测浓度的比值介于78.6%~95.1%之间,甲基丙烯酸甲酯的检测浓度与初始(0h)检测浓度的比值介于78.1%~94.9%之间,甲苯的检测浓度与初始(0h)检测浓度的比值介于75.7%~94.0%之间,二甲苯的检测浓度与初始(0h)检测浓度的比值介于77.5%~95.4%之间;二乙烯苯的检测浓度与初始(0h)检测浓度的比值介于70.4%~97.2%之间;苯乙烯的检测浓度与初始(0h)检测浓度的比值介于74.8%~95.5%之间;苯的检测浓度与初始(0h)检测浓度的比值介于76.5%~93.1%之间;1,2-二氯乙烷的检测浓度与初始(0h)检测浓度的比值介于72.1%~93.0%之间;100%限度浓度加标供试品溶液中正庚烷的检测浓度与初始(0h)检测浓度的比值介于74.7%~88.9%之间,乙醇的检测浓度与初始(0h)检测浓度的比值介于76.2%~90%之间,氯苯的检测浓度与初始(0h)检测浓度的比值介于74.5%~90.5%之间,甲基丙烯酸甲酯的检测浓度与初始(0h)检测浓度的比值介于78.3%~91.4%之间,甲苯的检测浓度与初始(0h)检测浓度的比值介于75.7%~90.5%之间,二甲苯的检测浓度与初始(0h)检测浓度的比值介于76.3%~91.0%之间;二乙烯苯的检测浓度与初始(0h)检测浓度的比值介于78.9%~88.6%之间;苯乙烯的检测浓度与初始(0h)检测浓度的比值介于75.9%~90.4%之间;苯的检测浓度与初始(0h)检测浓度的比值介于78.9%~90.5%之间;1,2-二氯乙烷的检测浓度与初始(0h)检测浓度的比值介于74.8%~89.4%之间;因此,供试品溶液、100%限度浓度对照品溶液和100%限度浓度加标供试品溶液于室温条件下放置至少28.5小时内稳定。
34.对比例溶液配制方法中空白溶液采用10% dmso,配制100%浓度对照品溶液,参照实施例的气相色谱条件参数和质谱条件参数,进样检测。检测结果见图2所示,结果表明,受预处理试剂影响,10种小分子待测物中正庚烷(10)未出峰,苯乙烯(8)的分离度不能达到要求,二乙烯苯(9)基线受干扰严重,导致检测方法不能用于红霉素样品中10种小分子物质的同时测定。
35.以上所述,以上实施例仅用以说明本技术的技术方案,而非对其限制;尽管参照前述实施例对本技术进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本技术各实施例技术方案的精神和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1