设计目标区域特异性液相探针的方法和系统与流程
本发明涉及设计目标区域特异性液相探针的方法和系统。
背景技术:
伴随测序技术的不断发展,在未来我们可以在较快的时间内以较为廉价的价格获得人类的基因组序列,对于dna的研究将更加便利。但在现阶段,想要获得一个人类基因组的dna测序序列,通常需要较为昂贵的价格,而且对于研究者而言,更多的针对某种疾病来研究可能引起该疾病的基因的dna序列的变异情况,对于研究者而言,全基因组测序一方面价格较高,另一方面存在有大量的冗余数据。因此如果可以通过某些技术来捕获期望的目标区域的dna片段,通过对捕获的目标dna片段进行测序,一方面节约经费,另一方面缩小测序范围,降低数据冗余。目标区域捕获测序技术应运而生,为生物研究带来了极大的便利。
目标区域捕获技术依托于探针。探针通常分为dna探针和rna探针,都表示一段与目的基因或dna(目标序列)互补的特异核苷酸序列。经过长期的发展,现阶段的探针由原来的20多bp逐步发展到现在的几十bp甚至上百bp的长度,不同的长度通常对应于不同的检测应用,对于人类基因组这类的区域捕获,通常探针长度在50bp以上。
目前主流的杂交捕获芯片有两种:on-arraycapture和in-solutioncapture,由于on-arraycapture有着较为明显的劣势,因此现在主流的杂交捕获芯片都是in-solutioncapture。对于in-solution,rochenimblegen和agilent是现阶段占据最大市场的份额的两家芯片提供商,用户通过提供感兴趣的目标区域给nimblegen和agilent,这两家公司提供相应的设计好的芯片反馈给用户。两家公司目前不提供开源的设计方式,用户只能得到设计好的芯片,无法知道芯片内具体的探针序列及对应的设计方式。目前国内也有不少生物芯片设计公司,但是大都是通过购买nimblegen和agilent的设计好的芯片来进行二次包装。
并且,目标区域的覆盖度,覆盖深度,均一性等这些对于研究者有着重要作用的因素,都是由选择的探针来决定的,因此探针设计方法对于商业芯片公司而言,是其重要竞争力。对于探针设计方法,可参考资料很少,尤其国内暂未见诸相关的文献的报道,寥寥可数的几款设计软件多为国外一些机构提供,而且这些机构通常提供相应的芯片服务,公众只能通过文献来大体了解其设计理念,无法获知具体的设计方法,从而,现阶段的学科研究和科学发展都急需一种公开的目标区域特异性液相探针设计方法。
技术实现要素:
本发明旨在至少解决现有技术中存在的技术问题之一。为此,本发明的一个目的在于提出一种能够快速、有效地设计目标区域特异性液相探针的手段。
根据本发明的一个方面,本发明提供了一种设计目标区域特异性液相探针的方法。根据本发明的实施例,该方法包括以下步骤:
(1)将目标区域向上下游延伸预定长度,以便获得经过处理的目标区域,所述经过处理的目标区域是由数量为wnum的窗口构成的;
(2)针对所述经过处理的目标区域,间隔固定步长选取长度为plen的探针序列,以便构建获得初始探针集;
(3)对所述初始探针集进行第一筛选,去除含有未知碱基的探针序列,以便获得经过第一筛选的探针集;
(4)确定所述经过第一筛选的探针集中每一条探针的探针属性参数;
(5)基于所述探针属性参数,对所述经过第一筛选的探针集进行第二筛选,以便获得经过第二筛选的探针集;
(6)确定所述经过第二筛选的探针集中每一条探针的分值pscore;
(7)针对所述经过处理的目标区域的每个窗口,基于所述经过第二筛选的探针集中在所述窗口内的所有探针的pscore值,确定一条最优探针;以及
(8)合并所有窗口的最优探针,以便获得最优探针集。
发明人惊奇地发现,利用本发明的方法能够快速、高效地设计获得目标区域的特异性液相探针,并且本发明的方法对目标区域的来源物种没有限制,对目标区域的数量也没有限制,且大小大于0bp的目标区域均适用于本发明的方法。此外,根据本发明的实施例,该方法尤其适合与深圳华大基因研究院的合成平台合成仪(例如oligoarray芯片式合成仪)配合使用,也即本发明的方法所采用的探针设计流程对应上述合成平台合成仪的探针制备策略。
根据本发明的另一方面,本发明还提供了一种用于设计目标区域特异性液相探针的系统。根据本发明的实施例,该系统包括:
区域延伸装置,所述区域延伸装置用于将目标区域向上下游延伸预定长度,以便获得经过处理的目标区域,所述经过处理的目标区域是由数量为wnum的窗口构成的;
初始探针集构建装置,所述初始探针集构建装置与所述区域延伸装置相连,用于针对所述经过处理的目标区域,间隔固定步长选取长度为plen的探针序列,以便构建获得初始探针集;
第一筛选装置,所述第一筛选装置与所述初始探针集构建装置相连,用于对所述初始探针集进行第一筛选,去除含有未知碱基的探针序列,以便获得经过第一筛选的探针集;
探针属性参数确定装置,所述探针属性参数确定装置与所述第一筛选装置相连,用于确定所述经过第一筛选的探针集中每一条探针的探针属性参数;
第二筛选装置,所述第二筛选装置与所述探针属性参数确定装置相连,用于基于所述探针属性参数,对所述经过第一筛选的探针集进行第二筛选,以便获得经过第二筛选的探针集;
pscore确定装置,所述pscore确定装置与所述第二筛选装置相连,用于确定所述经过第二筛选的探针集中每一条探针的分值pscore;
最优探针确定装置,所述最优探针确定装置与所述pscore确定装置相连,用于针对所述经过处理的目标区域的每个窗口,基于所述经过第二筛选的探针集中在所述窗口内的所有探针的pscore值,确定一条最优探针;以及
最优探针集构建装置,所述最优探针集构建装置与所述最优探针确定装置相连,用于合并所有窗口的最优探针,以便获得最优探针集。
根据本发明的实施例,利用本发明的系统能够快速、高效地设计获得目标区域的特异性液相探针,并且,该系统的适用于任何已测序的物种,对目标区域的数量和大小也没有特别限制,大于0bp的目标区域均可适用。此外,该系统尤其适合与深圳华大基因研究院的合成平台合成仪(例如oligoarray芯片式合成仪)配合使用。
根据本发明实施例的设计目标区域特异性液相探针的方法和系统具有下列优点的至少之一:
1、本发明的目标区域特异性液相探针设计技术快速有效,对于小于5mbp的目标区域,可在24小时内完成设计并反馈。
2、本发明的方法和系统,针对dna序列自身属性进行探针设计,可与深圳华大基因研究院合成仪配合使用,但不限于该合成仪,也即本发明具有一定的通用性。
3、本发明的方法和系统,能够保证目标区域的覆盖度,能够对目标区域进行评估,详细生成对目标区域能够覆盖情况以及覆盖的探针数量,并且能直观地获得生成覆盖目标区域的每一条探针的自身属性,用户可以依据生成的探针序列方便地调整覆盖的深度情况和理论覆盖度。
4、本发明的方法和系统,能够让使用人员依据实际情况灵活处理时间和空间的问题。具体地,本发明的技术方案是基于目标区域来进行设计的,用户可以灵活的将所有的区域一并进行设计也可以依据自身的计算性能分割区域进行设计,最终将所设计的结果合并即可。
本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
本发明的上述和/或附加的方面和优点从结合下面附图对实施例的描述中将变得明显和容易理解,其中:
图1显示了根据本发明一个实施例,探针设计方法中探针初始化阶段的流程示意图;
图2显示了根据本发明一个实施例,探针设计方法中目标区域探针选择阶段的流程示意图。
具体实施方式
下面详细描述本发明的实施例。下面描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。
需要说明的是,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括一个或者更多个该特征。进一步地,在本发明的描述中,除非另有说明,“多个”的含义是两个或两个以上。
方法
根据本发明的一个方面,本发明提供了一种设计目标区域特异性液相探针的方法。根据本发明的实施例,该方法包括以下步骤:
(1)将目标区域向上下游延伸预定长度,以便获得经过处理的目标区域,所述经过处理的目标区域是由数量为wnum的窗口构成的;
(2)针对所述经过处理的目标区域,间隔固定步长选取长度为plen的探针序列,以便构建获得初始探针集;
(3)对所述初始探针集进行第一筛选,去除含有未知碱基的探针序列,以便获得经过第一筛选的探针集;
(4)确定所述经过第一筛选的探针集中每一条探针的探针属性参数;
(5)基于所述探针属性参数,对所述经过第一筛选的探针集进行第二筛选,以便获得经过第二筛选的探针集;
(6)确定所述经过第二筛选的探针集中每一条探针的分值pscore;
(7)针对所述经过处理的目标区域的每个窗口,基于所述经过第二筛选的探针集中在所述窗口内的所有探针的pscore值,确定一条最优探针;以及
(8)合并所有窗口的最优探针,以便获得最优探针集。
发明人惊奇地发现,利用本发明的方法能够有效地设计获得目标区域的特异性液相探针,并且本发明的方法对目标区域的来源物种没有限制,对目标区域的数量也没有限制,且大小大于0bp的目标区域均适用于本发明的方法。此外,根据本发明的实施例,该方法尤其适合与深圳华大基因研究院的合成平台合成仪(例如oligoarray芯片式合成仪)配合使用,也即本发明的方法所采用的探针设计流程对应上述合成平台合成仪的探针制备策略。
根据本发明的实施例,在步骤(1)中,目标区域向上下游延伸的预定长度不受特别限制,实际操作中,可通过程序参数进行调整。根据本发明的一些具体示例,所述预定长度为30-50bp,优选40bp。
根据本发明的实施例,在步骤(2)中,所述固定步长为1-3bp,优选1bp。
根据本发明的实施例,在步骤(2)中,plen为30-200bp,优选90bp。
根据本发明的实施例,在步骤(4)中,在步骤(4)中,所述探针属性参数为选自探针序列中简单重复序列的含量prepeat、探针序列的15mer的频数的平均值pavgkmerfreq、探针序列在源基因组中的比对次数pwhits和探针序列是否含有连续相同的碱基pispoly的至少一种。由此,有利于探针的筛选,获得的探针质量高,特异性好。
根据本发明的实施例,prepeat是按照以下公式计算获得的:
其中,rep(p)为探针中的小写字符的数量,plen为探针的长度。
根据本发明的实施例,pavgkmerfreq是按照以下公式计算获得的:
其中,j=plen-15+1,f(i)为探针上第i个15mer的频数,plen为探针的长度。
根据本发明的实施例,所述连续相同碱基数量为8。由此,有利于探针的筛选,获得的探针质量高,特异性好。
根据本发明的实施例,在步骤(5)中,所述第二筛选进一步包括:
(a)判断探针序列的prepeat是否超过参数设定值,如果是则丢弃该探针;
(b)判断探针序列的pavgkmerfreq是否超过参数设定值,如果是则丢弃该探针;
(c)判断探针序列的pwhits是否超过参数设定值,如果是则丢弃该探针;
(d)判断探针序列的pispoly属性是否为真,如果是则丢弃该探针,其中,以探针序列含有连续相同的碱基表示pispoly属性为真,以探针序列不含有连续相同的碱基表示pispoly属性为假。由此,探针筛选效率高,效果好,获得的探针特异性好。
根据本发明的实施例,prepeat的参数设定值为0~1,pavgkmerfreq的参数设定值为1~255优选100,pwhits的参数设定值为大于1。由此,探针筛选效率高,效果好。
根据本发明的实施例,在步骤(6)中,pscore是按照以下公式计算获得的:
pscore=wwhits*pwhits+wtm*tmscore+wkmerfreq*kmerscore+wgc*gcscore+wrepeat*repscore,
其中,
wwhits+wtm+wkmerfreq+wgc+wrepeat=1,
pwhits为探针序列在源基因组中的比对次数,
wwhits为pwhits的权重系数,
tmscore是按照以下公式计算获得的:tmscore=-|ptm-tmexpc|/tmexpc,其中tmexpc为通过参数设置的预期的探针的解链温度,ptm为探针的解链温度,ptm=81.5+16.6(log10([na+]))+0.41*pgc-600/plen,[na+]表示钠离子的浓度,pgc表示探针的gc含量,plen为探针的长度,
wtm为tmscore的权重系数,
kmerscore是按照以下公式计算获得的:kmerscore=1/pavgkmerfreq,其中
wkmerfreq为kmerscore的权重系数,
gcscore是按照以下公式计算获得的:
wgc为gcscore的权重系数,
repscore是按照以下公式计算获得的:repscore=-prepeat,其中prepeat探针简单重复序列的含量,
wrepeat为repscore的权重系数。
根据本发明的实施例,在步骤(1)中,wnum是按照以下公式计算获得的:
wnum=targetsize*pdepth/plen,
其中,targetsize为目标区域的大小,pdepth为探针深度,plen为探针长度。
根据本发明的实施例,在步骤(7)中,针对所述经过处理的目标区域的每个窗口,将所述经过第二筛选的探针集里起始位置在所述窗口内的所有探针中pscore值最高的探针作为最优探针。
根据本发明的实施例,该方法进一步包括:
(9)确定所述最优探针集对所述目标区域的覆盖情况;以及
(10)基于所述最优探针集对所述目标区域的覆盖情况,进行如下操作:
a、当所述最优探针集覆盖了所述目标区域时,终止操作;
b、当所述目标区域中存在没有被所述最优探针集覆盖的区域,且没有被覆盖的区域达到45bp或者占所述目标区域大小的20%以上时,使用用户设置的更宽松的prepeat,返回重复步骤(4)-(8),对所述没有被覆盖的区域重新进行探针筛选,直到使用了用户允许的最大的prepeat为止。由此,能够保证目标区域的每个窗口均有探针覆盖,获得的探针质量高。
根据本发明的一些具体示例,prepeat是按照以下公式计算获得的:
其中,rep(p)为探针中的小写字符的数量,plen为探针的长度。
另外,根据本发明的实施例,本发明的方法还可以进一步包括“将所述经过处理的目标区域划分成数量为wnum的窗口”的步骤。且需要说明的是,该步骤的进行时间不受特别限制,本领域技术人员可以理解,可以在步骤(1)之后和步骤(7)之前的任何时间进行该步骤,只要方便在步骤(6)之后有效针对已划分好的数量为wnum的窗口分别确定最优探针即可。例如,可以在步骤(1)和步骤(2)之间,……,或者步骤(6)和步骤(7)之间进行。
此外,需要说明的是,本发明的方法依托于目标区域的源基因组序列(也称reference,即参考基因组序列)以及对应的目标区域,所有的探针序列均来源于源基因组。根据本发明的另一些实施例,本发明的设计目标区域特异性液相探针的方法分为两个阶段:探针设计初始化阶段和目标区域探针选择阶段。
下面分别详述这两个阶段:
i、探针设计初始化(参照图1)
在着手探针设计前,首先要确保源基因组和目标区域的匹配。例如对于人类基因组,本发明目前主要使用来自ncbi的grch37版本。但是根据用户需求,本发明也可以使用其他版本的源基因组。对于其他的物种,本发明要求提供相应的源基因组序列。本发明要求提供目标区域,目标区域数量不限定,大小要求大于0bp。本发明结合源基因组序列,扫描目标区域,并根据设置的参数对目标区域进行如下处理:
首先,对目标区域向两边延伸预定长度(即参数设定的长度)。
待目标区域经过上述步骤处理后,进入探针的初始化阶段,也即本发明对经过处理的目标区域,按照以下步骤进行探针初始化:
1)从源基因组中取得目标区域所在的dna碱基序列信息,以目标区域的起始作为起始位点,每隔固定步长选取特定长度(探针长度,下述记为plen)的碱基序列,直至最后一条碱基序列的起始位置加上plen超出目标区域的结束位置为止。
2)对于步骤1)中所获取的长度为plen的碱基序列,本发明认为该序列即为初始的探针序列。对于每一个探针p,通过染色体编号和探针的起始位置来唯一标识一条探针,在获得探针序列后,计算该探针的以下属性:
a)探针序列中是否含有未知碱基,未知碱基通常用‘n’表示。
b)探针序列的15mer的频数的平均值pavgkmerfreq。该属性用于检测探针的特异性,表征探针序列的交叉杂交的能力。15mer指长度为15bp的碱基序列。探针的平均频数的计算依赖于全基因组的15mer频数表,该表可以通过下述方式构建:假设全基因组总共含有g个碱基,通过逐个碱基滑动来截取15mer的方式,总共可以获得g-15+1个15mer,则可以构建这样一个表格,表格的第一列记录15mer的碱基序列信息,第二列记录该15mer在全基因组中出现的次数(即频数)。我们在长度为plen的探针上逐个碱基滑动来截取15mer,并到频数表中去查找每一个15mer的频数,然后计算探针序列的15mer的频数的平均值,计算公式如下:
其中,j=plen-15+1,f(i)为探针上第i个15mer的频数,plen为探针p的长度。
c)探针序列在源基因组中的比对次数pwhits。该属性同样用于表征探针的特异性。该数值越高表示探针和非目标区域结合的概率越大,特异性越差。本发明使用比对软件burrows-wheeleraligner(参见http://bio-bwa.sourceforge.net/)将探针序列比对回源基因组,获得探针在源基因组中的比对次数。
d)探针序列的gc含量pgc。碱基g和碱基c之间结合通常形成三价键,比a和t之间的二价键更加稳定。要形成稳定的gc结构,通常需要提供更多的能量,因此无论是探针的合成或者杂交捕获,gc含量越高的探针序列,通常越难以合成和捕获。探针gc含量的计算公式为:
其中,gc(p)为探针中的gc碱基的数量,plen为探针p的长度。
e)探针序列是否含有连续的相同的碱基pispoly。连续的相同的碱基序列会造成合成以及捕获的困难,尤其是连续的’g’和’c’。本发明扫描并检测探针是否含有8个以上的连续的相同的碱基。
f)探针序列的解链温度ptm。解链温度表示dna的双螺旋结构解开形成游离单链的温度。不同探针之间的解链温度的差异越小,捕获的效率越高。探针的解链温度的计算公式为:
ptm=81.5+16.6(log10([na+]))+0.41*pgc-600/plen,
其中,[na+]表示na离子的浓度,pgc表示探针的gc含量,plen为探针p的长度。
g)探针序列中的简单重复序列的含量prepeat,表征探针特异性。本发明把基因组序列上的小写字符[a,t,c,g]当成重复序列。当扫描探针序列的时候,可以通过计算探针序列内的小写字符来计算探针序列中的重复序列的含量,其计算公式为:
其中,rep(p)为探针中的小写字符的数量。plen为探针p的长度。
3)在步骤2)后,本发明对获得的探针进行初步的过滤。当某条探针序列含有未知碱基时,该探针会被过滤掉。
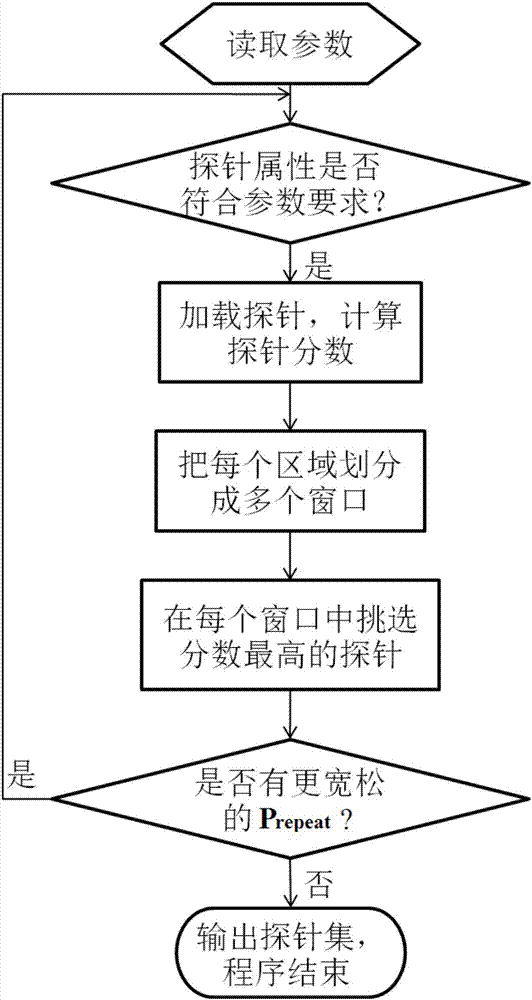
ii、目标区域探针选择(参照图2)
经过上述探针设计初始化阶段后,本发明对每个目标区域进行处理,在设置的参数要求下选择最优的探针。本发明的一个重要的特点在于可以实现多轮探针选择。在一次探针选择之后,对于目标区域中没有被探针序列覆盖的部分,本发明可以通过使用用户设置的不同的参数(主要是允许不同程度的prepeat)自动对这些没有被覆盖的部分进行新一轮的探针选择,以进一步提升探针序列对目标区域的覆盖度。探针选择的过程如下所述:
1)加载初始化探针集。加载过程中,进行以下处理:
a)判断探针序列的prepeat是否超过参数设定值,如果是则丢弃该探针;
b)判断探针序列的pavgkmerfreq是否超过参数设定值,如果是则丢弃该探针;
c)判断探针序列的pwhits是否超过参数设定值,如果是则丢弃该探针;
d)判断探针序列的pispoly属性是否为真,如果是则丢弃该探针。
2)加载完探针后,本发明通过以下公式计算每条探针的分值:
pscore=wwhits*pwhits+wtm*tmscore+wkmerfreq*kmerscore+wgc*gcscore+wrepeat*repscore,
其中各项的说明如下:
a)pwhits为探针序列在源基因组中的比对次数,wwhits为pwhits的权重系数。
b)tmscore为通过探针的解链温度ptm计算得到的分值,计算公式为:
tmscore=-|ptm-tmexpc|/tmexpc,
tmexpc为通过参数设置的预期的探针的解链温度,wtm为tmscore的权重系数。
c)kmerscore为通过探针的pavgkmerfreq计算得到的分值,计算公式为:
kmerscore=1/pavgkmerfreq,
wkmerfreq为kmerscore的权重系数。
d)gcscore为通过探针的gc碱基的数量gc(p)计算得到的分值,计算公式为:
其中,gcmin和gcmax分别为通过参数设置的gc碱基数量的最小值和最大值,gcmid=(gcmin+gcmax)/2。
e)repscore为通过探针的简单重复序列的含量prepeat计算得到的分值,计算公式为:
repscore=-prepeat,其中,prepeat探针简单重复序列的含量,
wrepeat为repscore的权重系数。
3)把每个目标区域划分成一定数量的窗口wnum,计算窗口数量的公式为:
wnum=targetsize*pdepth/plen,
其中,targetsize为目标区域的大小,pdepth为探针深度即覆盖目标区域中的每个碱基的探针的数量,plen为探针长度。
4)在每个窗口中,从起始位置在该窗口内的探针序列中选择分数最高的探针作为该窗口的最优探针。如果该最优探针的gc碱基的数量超出通过参数设置的gc碱基数量的最小值和最大值的范围,可根据参数设置对该最优探针进行拷贝,即在该窗口中使用两条一样的探针,以提高探针合成和捕获的成功率。
5)把每个窗口中的最优探针合并到一起即可得到一轮设计的最优探针集。
6)计算这一轮设计的最优的探针集对目标区域的覆盖情况。对于没有被覆盖的区域,如果其区域大小达到45bp,或者大小达到目标区域的大小的20%以上,则使用用户设置的更宽松的prepeat对这些区域进行探针挑选。重复1)到5)步,直到使用了用户允许的最大的prepeat为止。
在得到使用不同的prepeat进行挑选的探针集后,可供使用者根据项目目的选择某一个prepeat阈值下得到的最优探针集。
系统
根据本发明的另一方面,本发明还提供了一种适于实施前述的设计目标区域特异性液相探针的方法的系统。根据本发明的实施例,该用于设计目标区域特异性液相探针的系统包括:
区域延伸装置,所述区域延伸装置用于将目标区域向上下游延伸预定长度,以便获得经过处理的目标区域,所述经过处理的目标区域是由数量为wnum的窗口构成的;
初始探针集构建装置,所述初始探针集构建装置与所述区域延伸装置相连,用于针对所述经过处理的目标区域,间隔固定步长选取长度为plen的探针序列,以便构建获得初始探针集;
第一筛选装置,所述第一筛选装置与所述初始探针集构建装置相连,用于对所述初始探针集进行第一筛选,去除含有未知碱基的探针序列,以便获得经过第一筛选的探针集;
探针属性参数确定装置,所述探针属性参数确定装置与所述第一筛选装置相连,用于确定所述经过第一筛选的探针集中每一条探针的探针属性参数;
第二筛选装置,所述第二筛选装置与所述探针属性参数确定装置相连,用于基于所述探针属性参数,对所述经过第一筛选的探针集进行第二筛选,以便获得经过第二筛选的探针集;
pscore确定装置,所述pscore确定装置与所述第二筛选装置相连,用于确定所述经过第二筛选的探针集中每一条探针的分值pscore;
最优探针确定装置,所述最优探针确定装置与所述pscore确定装置相连,用于针对所述经过处理的目标区域的每个窗口,基于所述经过第二筛选的探针集中在所述窗口内的所有探针的pscore值,确定一条最优探针;以及
最优探针集构建装置,所述最优探针集构建装置与所述最优探针确定装置相连,用于合并所有窗口的最优探针,以便获得最优探针集。
发明人发现,利用本发明的系统能够有效地设计获得目标区域的特异性液相探针,并且,该系统的适用于任何已测序的物种,对目标区域的数量和大小也没有特别限制,大于0bp的目标区域均可适用。此外,根据本发明的实施例,该系统尤其适合与深圳华大基因研究院的合成平台合成仪(例如oligoarray芯片式合成仪)配合使用。
根据本发明的实施例,在所述区域延伸装置中,目标区域向上下游延伸的预定长度不受特别限制,实际操作中,可通过程序参数进行调整。根据本发明的一些具体示例,所述预定长度为30-50bp,优选40bp。
根据本发明的实施例,在所述初始探针集构建装置中,所述固定步长为1-3bp,优选1bp。
根据本发明的实施例,在所述初始探针集构建装置中,设plen为30-200bp,优选90bp。
根据本发明的实施例,所述探针属性参数为选自探针序列中简单重复序列的含量prepeat、探针序列的15mer的频数的平均值pavgkmerfreq、探针序列在源基因组中的比对次数pwhits和探针序列是否含有连续相同的碱基pispoly的至少一种。由此,有利于探针的筛选,获得的探针质量高,特异性好。
根据本发明的实施例,所述探针属性参数确定装置进一步包括:
prepeat确定单元,所述prepeat确定单元用于确定探针序列中简单重复序列的含量prepeat;
pavgkmerfreq确定单元,所述pavgkmerfreq确定单元用于确定探针序列的15mer的频数的平均值pavgkmerfreq;
pwhits确定单元,所述pwhits确定单元用于确定探针序列在源基因组中的比对次数pwhits;以及
pispoly属性确定单元,所述pispoly属性确定单元用于确定探针序列是否含有连续相同的碱基。
根据本发明的实施例,所述prepeat确定单元适于按照以下公式计算获得prepeat:
其中,rep(p)为探针中的小写字符的数量,plen为探针的长度。
根据本发明的实施例,所述pavgkmerfreq确定单元适于按照以下公式计算获得pavgkmerfreq:
其中,j=plen-15+1,f(i)为探针上第i个15mer的频数,plen为探针的长度。
根据本发明的实施例,在所述pispoly属性确定单元中,将所述连续相同碱基数量设置为8。由此,有利于探针的筛选,获得的探针质量高,特异性好。
根据本发明的实施例,所述第二筛选装置适于执行以下操作:
(a)判断探针序列的prepeat是否超过参数设定值,如果是则丢弃该探针;
(b)判断探针序列的pavgkmerfreq是否超过参数设定值,如果是则丢弃该探针;
(c)判断探针序列的pwhits是否超过参数设定值,如果是则丢弃该探针;
(d)判断探针序列的pispoly属性是否为真,如果是则丢弃该探针,其中,以探针序列含有连续相同的碱基表示pispoly属性为真,以探针序列不含有连续相同的碱基表示pispoly属性为假。由此,探针筛选效率高,效果好,获得的探针特异性好。
根据本发明的实施例,prepeat的参数设定值为0~1,pavgkmerfreq的参数设定值为1~255优选100,pwhits的参数设定值为大于1。由此,探针筛选效率高,效果好,获得的探针特异性好。
根据本发明的实施例,所述pscore确定装置适于按照以下公式计算获得pscore:
pscore=wwhits*pwhits+wtm*tmscore+wkmerfreq*kmerscore+wgc*gcscore+wrepeat*repscore
其中,
wwhits+wtm+wkmerfreq+wgc+wrepeat=1,
pwhits为探针序列在源基因组中的比对次数,
wwhits为pwhits的权重系数,
tmscore是按照以下公式计算获得的:tmscore=-|ptm-tmexpc|/tmexpc,其中tmexpc为通过参数设置的预期的探针的解链温度,ptm为探针的解链温度,ptm=81.5+16.6(log10([na+]))+0.41*pgc-600/plen,[na+]表示钠离子的浓度,pgc表示探针的gc含量,plen为探针的长度,
wtm为tmscore的权重系数,
kmerscore是按照以下公式计算获得的:kmerscore=1/pavgkmerfreq,其中
wkmerfreq为kmerscore的权重系数,
gcscore是按照以下公式计算获得的:
wgc为gcscore的权重系数,
repscore是按照以下公式计算获得的:repscore=-prepeat其中prepeat探针简单重复序列的含量,
wrepeat为repscore的权重系数。
根据本发明的实施例,按照以下公式确定构成所述经过处理的目标区域的窗口数量wnum:
wnum=targetsize*pdepth/plen,
其中,targetsize为目标区域的大小,pdepth为探针深度,plen为探针长度。
根据本发明的实施例,在所述最优探针确定装置中,针对所述经过处理的目标区域的每个窗口,所述最优探针为所述经过第二筛选的探针集里起始位置在所述窗口内的所有探针中pscore值最高的探针。
根据本发明的实施例,该系统进一步包括:
覆盖情况确定装置,所述覆盖情况确定装置与所述最优探针集构建装置相连,用于确定所述最优探针集对所述目标区域的覆盖情况;以及
检测装置,所述检测装置与所述覆盖情况确定装置相连,用于基于所述最优探针集对所述目标区域的覆盖情况进行如下操作:
a、当所述最优探针集覆盖了所述目标区域时,终止操作;
b、当所述目标区域中存在没有被所述最优探针集覆盖的区域,且没有被覆盖的区域达到45bp或者占所述目标区域大小的20%以上时,使用用户设置的更宽松的prepeat,返回所述探针属性参数确定装置,以便对所述没有被覆盖的区域重新进行探针筛选,直到使用了用户允许的最大的prepeat为止。由此,能够保证目标区域的每个窗口均有探针覆盖,获得的探针质量高。
根据本发明的一些具体示例,prepeat是按照以下公式计算获得的:
其中,rep(p)为探针中的小写字符的数量,plen为探针的长度。
另外,根据本发明的实施例,本发明的系统还可以进一步包括:窗口划分单元,所述窗口划分单元适于将所述经过处理的目标区域划分成数量为wnum的窗口。且需要说明的是,所述窗口划分单元的安装位置不受特别限制,本领域技术人员可以理解,可以设置于区域延伸装置、初始探针集构建装置、第一筛选装置、探针属性参数确定装置、第二筛选装置和pscore确定装置中的任意之一的内部,或者任意相连的两个装置之间,只要方便有效针对已划分好的数量为wnum的窗口分别确定最优探针即可。
下面将结合实施例对本发明的方案进行解释。本领域技术人员将会理解,下面的实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件(例如参考j.萨姆布鲁克等著,黄培堂等译的《分子克隆实验指南》,第三版,科学出版社)或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品,例如可以采购自illumina公司。
一般方法:
本发明实施例的设计目标区域特异性液相探针的一般方法,包括以下步骤:
(1)将目标区域向上下游延伸预定长度,以便获得经过处理的目标区域;
(2)针对所述经过处理的目标区域,间隔固定步长选取长度为plen的探针序列,以便构建获得初始探针集;
(3)对所述初始探针集进行第一筛选,去除含有未知碱基的探针序列,以便获得经过第一筛选的探针集;
(4)确定所述经过第一筛选的探针集中每一条探针的探针属性参数;
(5)基于所述探针属性参数,对所述经过第一筛选的探针集进行第二筛选,以便获得经过第二筛选的探针集;
(6)确定所述经过第二筛选的探针集中每一条探针的分值pscore;
(7)将所述经过处理的目标区域划分成数量为wnum的窗口;
(8)针对每个窗口,基于所述经过第二筛选的探针集中在所述窗口内的所有探针的pscore值,确定一条最优探针;
(9)合并所有窗口的最优探针,以便获得最优探针集;
(10)确定所述最优探针集对所述目标区域的覆盖情况;以及
(11)基于所述最优探针集对所述目标区域的覆盖情况,进行如下操作:
a、当所述最优探针集覆盖了所述目标区域时,终止操作;
b、当所述目标区域中存在没有被所述最优探针集覆盖的区域,且没有被覆盖的区域达到45bp或者占所述目标区域大小的20%以上时,使用用户设置的更宽松的prepeat,返回重复步骤(4)-(9),对所述没有被覆盖的区域重新进行探针筛选,直到使用了用户允许的最大的prepeat为止。
发明人参照前述的“一般方法”(其中涉及的各公式的计算参照前面的具体实施方式),针对人类基因组(使用来自ncbi的grch37版本)的目标区域进行探针设计,以下为两个具体实施例。
实施例1:
1)背景
用户提供的目标区域的大小86,125bp,共包含274个目标区域,具体的目标区域如下表所示:
2)设计过程
使用的设计参数为:--targettarget.bed--outdir.--usrprojexample--extl20--depth42>coverage.txt,
其中target指定包含目标区域的bed格式的文件,outdir指定输出结果的路径,usrproj指定项目名称,extl指定目标区域两边延伸的长度,depth为预期的探针深度,其余参数均为程序默认值。
3)设计结果
该项目设计需要的时间为3分钟。
4)实验结果
使用两个样本进行捕获实验的结果如下表所示。
实施例2
1)背景
用户提供的目标区域的大小164,999bp,共包含376个目标区域,具体的目标区域如下表所示:
2)设计过程
该项目进行了两步设计。第一步设计使用的设计参数为:--targettarget.bed--outdir.--usrprojexample--depth4--expectl70,
其中target指定包含目标区域的bed格式的文件,outdir指定输出结果的路径,usrproj指定项目名称,depth指定探针的深度,expectl为期望的探针长度,其余参数均为程序默认值。
在第一步设计的基础上,对于两个没有被覆盖的较大的区域,使用以下参数进行第二步设计:--targetuncovered.bed--outdir.--usrprojexample--depth4--expectl70--homohits4,
其中,homohits指定允许探针比对到参考序列上的不同位置的数量。
uncovered.bed包括的区域为:
chr9135937314135937505
chr9135944058135944646
3)设计结果
第一步设计的结果如下表所示。
第二步设计的结果如下表所示。
完成两步设计后,把两步设计的探针序列合并得到最终的探针序列集。
该项目设计需要的时间为11分钟。
4)实验结果
使用两个样本进行捕获实验的结果如下表所示。
上述两个实施例体现探针设计结果有着良好的覆盖度,并且提供了不同参数(探针最大重复序列比例)下的设计结果,可以让用户根据项目需要进行选择,有较好的灵活性。而且项目设计需要的时间较短,允许用户在有限的时间内尝试更多不同的参数进行设计,以获得更好的设计结果。实验结果显示,上述两个实施例的目标区域的覆盖率达到98%以上,捕获效率(即比对到目标区域数据比例)分别为64%左右和42%左右,均达到市场主流商业探针的水平。
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。
尽管已经示出和描述了本发明的实施例,本领域的普通技术人员可以理解:在不脱离本发明的原理和宗旨的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由权利要求及其等同物限定。
- 还没有人留言评论。精彩留言会获得点赞!