预测肺癌病人预后的基因表达分类器及其构建方法与流程
本发明涉及一种基因表达分类器及其构建方法,更具体地涉及一种预测肺癌病人预后的基因表达分类器及其构建方法,特别是一种预测非小细胞肺癌病人预后的基因表达分类器及其构建方法。
背景技术:
中国最新癌症流行病学调查显示2015年新增肺癌病人73万,死亡病人60万,肺癌已成为中国发病率和死亡率均处于首位的癌症(1)。肺癌死亡例数占总体癌症死亡例数的25%。由于肺癌的高复发风险和低存活率,绝大多数处于ib-iiia期的肺癌病人都会进行术后化疗(postoperativechemotherapy,poct)。此外,基于病灶残留程度、淋巴结转移情况、癌症分期等参数,相当一部分的肺癌病人也会进行术后放疗(postoperativeradiotherapy,port)(2-4)。人类癌症具有高度异质性,即使临床上被诊断为同一分期的癌症病人对于同样的治疗方案的反应和总体预后也会大不相同(5)。换言之,有相当一部分病人不能从辅助疗法中获得生存益处或者只能获得很少的生存益处,却要忍受不必要的放化疗痛苦(6-8)。近年来,科学家们一直尝试基于癌症的基因表达特征来开发相应的分子标记物和分子分类手段。在基因表达分类器领域,乳腺癌开始最早,进展最大,已有多个分子检测产品问世,如oncotypedx(9,10)、mammaprint(11,12)、prosigna(13,14)、endopredict(15)以及breastcancerindex(16)。其中,oncotypedx(又称乳腺癌21基因检测)与mammaprint这两个检测产品发展最为成熟,接受度最广,oncotypedx检测作为指导早期浸润性乳腺癌的术后辅助治疗方案选择的重要依据,已经被写入美国国立综合癌症网络(nccn)指南。到目前为止,也有研究尝试在肺癌领域开发类似的基因表达分类器来预测肺癌病人的复发风险(17-29),这些研究几乎都是针对非小细胞肺癌,但是存在以下几种问题:一是不同人群肺癌的生物异质性,从此人群和彼人群得出的基因表达分类器差异很大;二是缺乏统一的标准,如临床样本的采集、注释、样本处理等;三是统计和机器学习的方法千差万别,这些分类器涉及的基因数目千差万别,没有什么基因重合,且就哪个是最佳基因分类器也没有形成共识,因此未开展有影响力的临床验证研究。在本研究中,我们对tcga中肺腺癌(lungadenocarcinoma,adc)病人的rna-seq数据运用有监督的机器学习的方法设计得到一种基因表达分类器,实现对非小细胞肺癌病人的无复发生存期(relapse-freesurvival,rfs)和总体生存期(overallsurvival,os)进行精准预测。
技术实现要素:
一方面,本发明的目的是提供一种预测肺癌病人预后的基因表达分类器的构建方法,包括:数据训练阶段和验证阶段,所述训练阶段包括第一阶段和第二阶段,所述第一阶段使用有监督的机器学习方法建立能预测肺癌病人预后的基因表达分类器雏形,所述第二阶段进一步使用机器学习的方法获得预测肺癌病人预后的基因表达分类器。
另一方面,本发明的目的是提供一种预测肺癌病人预后的基因表达分类器的构建方法,其中所述第一阶段通过使用肺癌病人的基因表达信息和临床信息,采用有监督的机器学习方法挑选与真实预后情况高度相关的基因。
另一方面,本发明的目的是提供一种预测肺癌病人预后的基因表达分类器的构建方法,其中对所有基因的表达和预后情况的pearson系数的绝对值由大到小排序,获得与真实一年内复发高度相关的基因。
另一方面,本发明的目的是提供一种预测肺癌病人预后的基因表达分类器的构建方法,其中使用loocv方法获得最佳的基因类别和数目,从而构建所述基因表达分类器雏形。
另一方面,本发明的目的是提供一种预测肺癌病人预后的基因表达分类器的构建方法,其中使用基因表达分类器雏形计算用于验证的肿瘤样本的风险系数,预测其复发风险,通过比较真实复发风险和预测复发风险的一致性,验证所述基因表达分类器雏形的效能。
另一方面,本发明的目的是提供一种预测肺癌病人预后的基因表达分类器的构建方法,其中第一阶段的基因表达分类器雏形包含有基因列表。
另一方面,本发明的目的是提供一种预测肺癌病人预后的基因表达分类器的构建方法,其中所述第二阶段包括特征排序,所述特征排序基于单因素的cox比例风险回归模型所得p值进行。
另一方面,本发明的目的是提供一种预测肺癌病人预后的基因表达分类器的构建方法,其中所述第二阶段采用km生存分析计算高风险组和低风险组之间的时序检验p值,时序检验p值最小的cox模型所包含的基因种类和数目就是所构建的基因表达分类器。
另一方面,本发明的目的是提供一种预测肺癌病人预后的基因表达分类器的构建方法的构建方法,其中验证阶段使用芯片数据集验证所获得的基因表达分类器。
另一方面,本发明的目的是提供一种预测非小细胞肺癌病人预后的基因表达分类器及其构建方法。
另一方面,本发明的目的是提供一种预测非小细胞肺腺癌病人预后的基因表达分类器及其构建方法。
另一方面,本发明的目的是提供一种预测非小细胞肺鳞癌病人预后的基因表达分类器及其构建方法。
另一方面,本发明的目的是提供一种预测非小细胞肺腺癌和肺鳞癌病人预后的基因表达分类器及其构建方法。
另一方面,本发明的目的是提供一种预测非小细胞肺癌病人预后的基因表达分类器及其构建方法,实现对非小细胞肺癌病人的无复发生存期和总体生存期进行精准预测。
另一方面,本发明的目的是提供一种预测非小细胞肺癌病人预后的基因表达分类器及其构建方法,将非小细胞肺癌病人分为差预后和预后良好的两个亚群。
另一方面,本发明的目的是提供一种在多个非小细胞肺癌数据集中预测非小细胞肺癌病人预后的基因表达分类器及其构建方法。
另一方面,本发明的目的是提供一种在多个非小细胞肺癌数据集中预测非小细胞肺癌病人预后的基因表达分类器及其构建方法,所述多个非小细胞肺癌数据集选自由tcga、gse8894、gse31210、gse11969、gse13213、gse14814或gse37745。
另一方面,本发明提供一种预测非小细胞肺癌病人预后的基因表达分类器的构建方法,其包括:数据训练阶段和验证阶段。
另一方面,本发明提供一种预测非小细胞肺癌病人预后的基因表达分类器的构建方法,其中所述训练阶段包括第一阶段和第二阶段。
另一方面,本发明提供一种预测非小细胞肺癌病人预后的基因表达分类器的构建方法,其中所述第一阶段包括:使用未接受术后放疗的tcga肺腺癌病人的基因表达信息和临床信息,使用有监督的机器学习方法建立能预测肺腺癌病人预后的基因表达分类器雏形。
另一方面,本发明提供一种预测非小细胞肺癌病人预后的基因表达分类器的构建方法,其中有监督的机器学习的方法如下:
分组:对满足条件的非小细胞肺腺癌病人的基因表达数据在分组前进行标准化处理,然后分别从临床上差预后和预后良好的病人中随机选取第一数量病人和第二数量作为训练组,剩余病人作为验证组;
挑选与真实预后情况高度相关的基因:初始rna-seq数据经过初步过滤,从多个基因中获得在病人中表达量不为0的基因;在训练病人组内,每个基因的表达量与这些病人的真实预后情况进行pearson相关分析,并获得回归系数(coef),|coef|≥0.3的基因被挑选出来进行下一步分析;
有监督的分类方法:将挑选出来的基因按照相关系数的绝对值从大到小进行排序,得到一个排行榜,从排在最前面的两个基因开始,每次从排行榜再依次添加两个基因建立一个分类器,如此循环,直到排行榜中所有的基因都被作为报告子用尽,建立基因表达分类器雏形。
另一方面,本发明提供一种预测非小细胞肺癌病人预后的基因表达分类器的构建方法,其中使用loocv检查所述基因表达分类器雏形的效能,步骤如下:
第一步,将一名病人留出,以备后续检验用;
第二步,通过取表达的标准分数的平均值计算在差预后组该分类器中所涉及的所有基因的表达模式;类似地计算在预后良好组的基因表达模式;接着,定义一个风险系数(risk-coef),所述风险系数即指该肿瘤内预后良好组的基因表达模式的相关系数减去差预后组基因表达模式的相关系数;
第三步,计算剩余的训练肿瘤样本以及第一步中留存的肿瘤样本的风险系数,将这些样本按照风险系数从小到大排序,第一数量肿瘤病人被划分为高基因组风险组,第二数量病人被划分为低基因组风险,检查每个病人真实的临床预后情况和预测的基因组风险的一致性;
循环第一到第三步,直到所有训练病人样本都被留出过一次,每次当被留出的样本的基因组风险和实际的临床预后情况相背离时,错误计数器加1次;
对于每一个分类器,设置风险系数的门槛值,将病人分为第一数量高风险和第二数量低风险的病人;同时,计算独立于训练样本的验证样本的风险系数;通过前面设置的风险系数的门槛值,可判定验证样本的基因组风险的高低;同样地,也统计验证组病人的基因组风险和实际临床预后出现不一致的次数;最终发现分别包含排行榜的前80或者前84个基因的分类器预测错误数目为最低,选择80-基因表达分类器作为雏形。
另一方面,本发明提供一种预测非小细胞肺癌病人预后的基因表达分类器的构建方法,其中运用一种改良版的loocv来解决信息遗露(informationleakage)的问题:
第一步,留出一个样本待验证;
第二步,运用剩余样本计算所有基因的表达和预后情况的pearson系数,过滤得到|coef|≥0.3的基因;
第三步,运用第二步中过滤得到的基因构建分类器,并据此预测被留出的那个样本的基因组风险。
第四步,重复第一到第三步,直到所有肿瘤病人都被留出过一次,从而获得基因表达分类器。
另一方面,本发明提供一种预测非小细胞肺癌病人预后的基因表达分类器的构建方法,其中所述第二阶段包括:
基于训练的第一阶段获得的80-基因表达分类器,进一步运用机器学习的方法获得更加简洁的风险评分系统来预测肺癌病人的预后,同样是运用tcga的病人数据,但是这次囊括了所有未接受和接受了术后放疗的病人,在建模过程中,基因被称为特征;
随机分组:将这些样本随机划分为:组1和组2。
特征排序:以组1为训练数据,通过单因素的cox比例风险回归模型计算单个特征的回归系数和p值,按照p值从小到大将80个特征重新排序,排在越前面的特征,cox回归p值越小,与预后的相关性越大;
特征数目的优化:从排序后的第一个特征开始,从前往后每次加一个特征,运用多因素的cox回归分析获得各个特征的cox回归系数;运用组2病人进行交叉验证,评估此cox模型的好坏:将组2中每个病人的相关特征的表达值与多因素cox回归系数相乘并累加得到一个分数值,分数值高低表示病人死亡或者复发风险的高低;接着采用km生存分析计算组2的高风险和低风险亚组之间的时序检验p值;如此循环,直到所有的特征都被纳入cox回归模型;时序检验p值最小的cox模型所包含的特征种类和数目就是最优的,获得基因表达分类器。
另一方面,本发明提供一种预测非小细胞肺癌病人预后的基因表达分类器的构建方法,其中所述验证阶段包括:
使用geo数据库内符合条件的非小细胞肺癌的芯片数据集验证以上所获得基因表达分类器;在一个独立的数据集中,那些分数值高于群体分数值中位数的病人被划为高风险组,而其余为低风险组;km分析用来比较高风险组和低风险组的生存曲线;时序检验p值<0.05表示有统计学差异。
在另一实施方式中,本发明提供一种19-基因表达分类器。
在另一实施方式中,本发明提供一种19-基因表达分类器,其中19基因表达分类器的基因选自由galnt2、c17orf50、stc2、c8orf46、znf441、znf563、znf763、tmem63c、znf442、c9orf135、actn1、c4orf12、csf2、znf879、nags、c15orf63、znf799、c6orf176、c14orf129组成的组。
另一方面,本发明提供一种基因表达分类器,其能作为一种有效的诊断手段将肺癌病人中有较高风险发展成差预后的亚群分离出来。
另一方面,本发明提供一种基因表达分类器,其能作为一种有效的诊断手段将非小细胞肺癌病人中有较高风险发展成差预后的亚群分离出来。
有益效果
本发明运用有监的机器学习的方法建立了一种基因表达分类器来精准预测肺癌预后。该基因表达分类器具有以下几种明显的优点:
既能预测肺腺癌又能预测肺鳞癌的预后。虽然第一阶段的80-基因表达分类器雏形是用来预测肺腺癌病人的复发风险的,然而最终版本的基因表达分类器却能同时预测肺腺癌和肺鳞癌(lungsquamouscellcarcinoma,scc)病人的总体生存期和无复发生存期,这证明该基因表达分类器的功能多样性。
预测功效非常强。我们在分类器的验证阶段用的是基因芯片表达数据集,这些芯片来自于不同的版本,包括gpl570、gpl7015、gpl6480以及gpl96。这些芯片平台中,并不是都能找到相应的探针来分别对应分类器中的19个基因。在gpl570、gpl7015、gpl6480以及gpl96中,19个基因中分别仅有17、9、12和6个基因可以找到相应的探针。因此在利用这些平台的基因芯片数据集进行验证时,我们只能提取17、9、12或6基因的表达值进行加权相加。出乎意料的是,这些所谓的“不完整的分类器”依然表现出非常强的预测功效。
可作为独立的非小细胞肺癌预后(总体生存期和无复发生存期)强诊断因子。我们的基因表达分类器的预后预测效能显著优于包括年龄、吸烟历史、基因突变、基因拷贝数变异等在内的临床诊断因子。另外,我们的基因表达分类器在不同的gse数据集中的预测效能都显著优于一个已经发表的16-基因分类器,由此可见该基因表达分类器的优越性。
附图说明
本发明的进一步特征和优点将结合附图进行描述,其中:
图1是所有基因的表达与一年内复发情况的相关系数分布图。(a)淡绿色:所有基因的基因表达和预后类别之间的相关系数分布,有1470个基因示出了相关程度大于0.3;淡红色分布是monte-carlo试验,其中对基因表达和预后类别之间的相关性进行随机化;(b):在10,000个monte-carlo试验中,|coef|≥0.3的基因数目的频数分布。
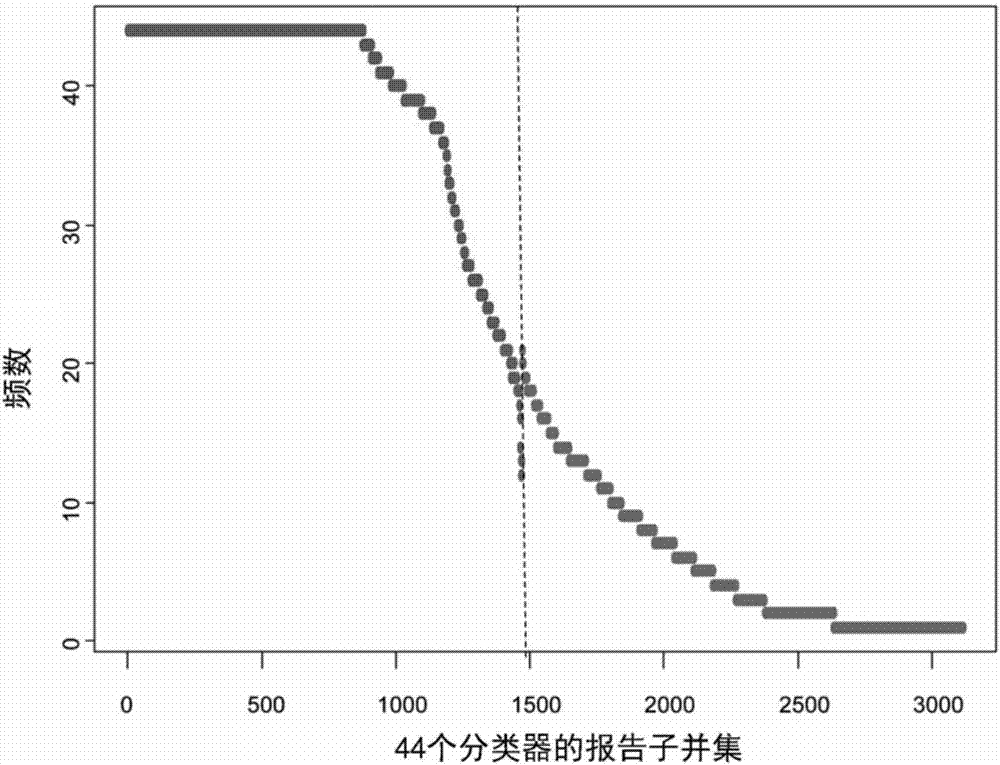
图2是以无信息遗露(informationleakage)的方式产生的44个基因表达分类器中,原初的1470个基因(红色)和其它基因(蓝色)的分布示意图。在一个基因表达分类器中,报告基因的平均数目是1523±98。
图3是80-基因表达分类器雏形的开发流程图。a-b:优化分类器雏形中的基因数目。a:在loocv循环中观察到的分类错误。x轴是分类器中报告子(基因)的数目;y轴是预测错误次数。采用前22、30、34、36、38、40、42、44、46、48、50、80、84和86个基因的分类器分别实现了最少的错误次数。b:利用额外的包含13个肿瘤的测试集验证上述的分类器的性能。80-基因和84-基因分类器实现了最低的错误次数。c:训练数据集内44个病人(上图)和测试数据集内13个病人(下图)的80个基因表达热图。每排代表一个观察(病人),每行是一个基因,基因名称标注在上图和下图之间。根据良好预后组和差预后组的平均表达模式的相关性对肿瘤进行排序(左图)。根据基因与两种预后类别的相关系数对基因进行排序。每个病人的真实预后状态在中间图中示出。黑色实线是80-基因表达分类器的系数阈值。
图4是tcga肺腺癌病人总体生存期(a)和无复发生存期(b)的高19-基因风险评分病人和低19-基因风险评分病人的km分析。
图5是geo数据集中病人的总体生存期的高19-基因风险评分和低19-基因风险评分的km分析(a-f)。
图6是geo数据集中病人的无复发生存期的高19-基因风险评分和低19-基因风险评分的km分析(a-d)。
图7在多变量cph模型中,19-基因表达评分优于其它临床病理因子和已公开的16-基因表达分类器(a-d)。a:gse31210肺腺癌总体生存期的多变量cph分析;b:gse31210肺腺癌无复发生存期的多变量cph分析;c:gse13213肺腺癌总体生存期的多变量cph分析;和(d)gse11969肺腺癌总体生存期的多变量cph分析。e和f:使用双变量cph模型比较本发明的19-基因表达分类器和已在nejm公开的16-基因表达分类器的预后值(e:总体生存期;e:无复发生存期)。横线表示95%置信区间,*、**和***分别表示p<0.05、p<0.01和p<0.001。
具体实施方式
本发明将会参照下面的实施例进行阐述,但本发明将不限于下面的实施例。
1.研究材料和手段
tcga和geo数据集
tcga的非小细胞肺癌病人rna-seq转录组数据及其临床信息从tcgarna-seq数据库中获得(https://cancergenome.nih.gov/)(表1)。而非小细胞肺癌病人的芯片表达数据及其临床信息从高通量基因表达(geneexpressionominibus,geo)数据库中获得(https://www.ncbi.nlm.nih.gov/geo/)(表1)。
表1:研究中使用的数据集
备注:adc:肺腺癌;scc:肺鳞癌;rfs:无复发生存期;os:总体生存期
基因表达分类器的开发流程
整个开发流程包括数据训练和验证两大阶段。
训练阶段
1.第一阶段
未接受术后放疗的tcga肺腺癌病人的基因表达信息和临床信息被用来建立能预测肺腺癌病人预后的80-基因的分类器雏形。该有监督的机器学习的方法如下:
分组
满足条件的非小细胞肺癌肺腺癌病人共57位,其中39位病人在术后一年之内发生了复发事件而被标记为临床上差预后的病人,而18位病人在术后至少大于一年的期间内未发生复发事件,被标记为临床上预后良好的病人。这些病人的基因表达数据在分组前进行标准化处理(z-评分)。我们分别从临床上差预后和预后良好的病人中随机选取了15和29名作为训练组。剩余的13例病人作为验证组。
挑选与真实预后情况高度相关的基因
初始rna-seq数据经过初步过滤,从共20530个基因中获得19574个基因在病人中表达量不为0。在训练病人组内,每个基因的表达量与这些病人的真实预后情况进行pearson相关分析,并获得回归系数(coef),|coef|≥0.3的基因(共1470个基因)被挑选出来进行下一步分析。为了排除获得的回归系数分布是随机概率产生的,我们运用一种排列(permutation)方法产生了10,000个monte-carlo模拟试验,从而对训练组的44个病人的基因表达数据和预后分组信息进行随机化处理。在10,000个monte-carlo试验中,|coef|≥0.3的基因数目的试验的频数分布见图1b。获得|coef|≥0.3的基因数目≥1470的概率为0.047(p<0.05),据此可拒绝零假设。
有监督的分类方法
很显然,1470个基因对于分类器来说数目太过庞大,基因数目需要优化。将这1470个基因按照相关系数的绝对值从大到小进行排序,得到一个排行榜(toplist)。从排在最前面的两个基因开始,每次从排行榜再依次添加两个基因建立一个分类器,如此循环,直到排行榜中所有的1470个基因都被作为报告子(reporters)用尽,因此总共建立了735个分类器。
一种称为loocv(leave-one-outcross-validation)的方法被用来检查这些分类器的效能,步骤如下:
第一步,将一名病人留出,以备后续检验用。
第二步,计算在差预后组该分类器中所涉及的所有基因的表达模式(poor-prognosisexpressiontemplate)(取表达z-评分的平均值);类似地计算在预后良好组的基因表达模式(good-prognosisexpressiontemplate)。接着,定义一个风险系数(risk-coef)。对于一个肿瘤来说,风险系数即指该肿瘤内预后良好组的基因表达模式的pearson相关系数减去差预后组基因表达模式的pearson相关系数:
风险系数=预后良好组的基因表达模式的pearson相关系数-差预后组基因表达模式的pearson相关系数
第三步,计算剩余的43个训练肿瘤样本以及第一步中留存的肿瘤样本的风险系数。将这44个样本按照风险系数从小到大排序,前面15个肿瘤病人被划分为高基因组风险(highgenomicrisk)组,而剩余的29个病人被划分为低基因组风险(lowgenomicrisk)组。检查每个病人真实的临床预后情况和预测的基因组风险的一致性。
循环第一到第三步,直到所有的44个训练病人样本都被留出过一次。每次当被留出的样本的基因组风险和实际的临床预后情况相背离时,错误计数器加1次。
最好的分类器在loocv过程中,错误计数器收集的预测错误次数应该最少。当我们的基因表达分类器包含排行榜的前22、30、34、36、38、40、42、44、46、48、50、80、84或86个基因时,loocv过程中的预测错误为0。还需要进一步判断这14个分类器中哪一个最好。对于每一个分类器,我们设置风险系数的门槛值,将44个病人分为15个高风险和29个低风险的病人。同时,我们计算独立于训练样本的13个验证样本的风险系数。通过前面设置的风险系数的门槛值,我们可判定这13个验证样本的基因组风险的高低。同样地,我们也统计验证组病人的基因组风险和实际临床预后出现不一致的次数。最终发现分别包含排行榜的前80个基因或者前84个基因的分类器预测错误数目为最低。本着简洁的原则,最终选择80-基因为分类器雏形。
无信息遗漏(informationleak)的交叉验证
由于以上1470个基因是基于所有的44个训练样本获得的,包括被留出的用来验证的那个样本,因此存在信息遗漏所产生的过度拟合的可能性。为此,我们运用一种改良版的loocv来解决信息遗漏的问题:
第一步,留出一个样本待验证。
第二步,运用剩余的43个样本计算所有基因的表达和预后情况的pearson系数。过滤得到|coef|≥0.3的基因。
第三步,运用第二步中过滤得到的基因构建分类器,并据此预测被留出的那个样本的基因组风险。
第四步,重复第一到第三步,直到所有的44个肿瘤病人都被留出过一次。
因此我们又获得44个分类器。我们发现原初的1470个基因中绝大多数的基因都存在于这44个分类器中(图2)。据此,我们判定前面的训练过程中所引入的信息遗漏相当有限。
2.第二阶段
基于训练的第一阶段获得的80-基因分类器,我们进一步运用机器学习的方法获得更加简洁的风险评分系统来预测肺癌病人的预后。同样是运用tcga的病人数据,但是这次囊括了所有未接受和接受了术后放疗的病人,这样总共有350个肺腺癌样本可用。关注癌症病人的无复发生存期(relapse-freesurvival,rfs)。在建模过程中,基因被称为特征(features)。
随机分组
将这些样本随机划分为(随机化后两组病人各临床信息无显著差异):组1和组2。
特征排序
以组1为训练数据,通过单因素的cox比例风险回归模型(cox’sproportionalhazardsregressionmodel,cph),计算单个特征的回归系数和p值。按照p值从小到大将80个特征重新排序。排在越前面的特征,cox回归p值越小,与预后的相关性越大。
特征数目的优化:这是一种迭代优化的过程。从排序后的第一个特征开始,从前往后每次加一个特征,运用多因素的cox回归分析获得各个特征的cox回归系数。运用组2病人进行交叉验证,评估此cox模型的好坏:将组2中每个病人的相关特征的表达值与多因素cox回归系数相乘并累加得到一个分数值(score),分数值高低表示病人死亡或者复发风险的高低。接着采用km(kaplan-meier)生存分析计算组2的高风险和低风险亚组之间的时序检验p值(logrankp-value)。如此循环,直到所有的特征都被纳入cox回归模型。时序检验p值最小的cox模型所包含的特征种类和数目就是最优的。
验证阶段
geo数据库内符合条件的非小细胞肺癌病人的芯片数据集被用来验证以上所获得19-基因分类器。在一个独立的数据集中,那些分数值高于群体分数值中位数的病人被划为高风险组,而其余为低风险组。km分析用来比较高风险组和低风险组的生存曲线。时序检验p值<0.05表示有统计学差异。
基因表达分类器和其他临床诊断因子在诊断预测性能的优劣比较
我们利用多因子的cox回归模型来比较该基因表达分类器和其他临床诊断因子,包括年龄、肿瘤分期、吸烟历史、基因突变、myc拷贝数变异等在肺癌病人预后预测性能方面的强弱。在进行分析之前,所有这些因子都转化为二分变量。某个因子的风险比(hazardratio,hr)是其cox回归系数的自然指数。p<0.05表示该因子可作为一个独立的诊断因子预测肺癌病人的预后。
结果
80-基因表达分类器的构建
从tcga肺腺癌rna-seq数据出发,我们通过一种无偏的筛选方法获得与病人预后显著相关的1470个基因(|coef|>=0.3)。接着在loocv过程中,我们依次获得735个分类器,并且当分类器包含排行榜的前22,30,34,36,38,40,42,44,46,48,50,80,84或86个基因的时候,loocv过程中预测错误计数为0(图3a)。进一步的验证,我们发现这14个分类器对44个训练组病人的预测准确率为100%。我们还留有13个独立于训练组的病人用于验证。评判一个分类器的好坏在于其误将真实的预后差的病人错误预测为低风险组的次数最少,结果发现80-基因分类器和84-基因分类器的错误预测次数最少,因此80-基因和84-基因分类器拥有最强的效能来准确预测肺腺癌病人在一年内的复发风险。为了简洁起见,最终选择80-基因进行下面的研究(图3c),相应的风险系数门槛值为-0.38。
19-基因表达分类器的构建
基于简洁有效的出发点,我们采取进一步的机器学习方法对80-基因分类器进行基因数目的优化,获得19-基因表达分类器,每个基因都有相应的表达回归系数(又称加权系数),因此每个病人的这19个基因的表达值进行加权相加,即得该病人的预后风险值(表2)。km生存分析显示在tcga肺腺癌病人中,高风险组病人的预后要显著差于低风险组病人的预后(os:cphhr=1.77,p=0.001,km时序检验p值=0.00091;rfs:cphhr=1.77,p=0.004,km时序检验p值=0.00334)(图4)。这与我们的预期相符合,因为19-基因分类器是利用相同的tcga病人数据开发得来的。
表2.通过19-基因风险分数计算计算19-基因的cox系数
19-基因表达表达分类器在其他非小细胞肺癌数据集中的验证
该19-基因分类器的验证需要在数个独立于机器学习过程中的非小细胞肺癌数据集内进行。我们从geo中获得多个可用的基于基因芯片表达的数据集(表1)。就总体生存期指标而言,19-基因表达分类器可以成功地将gse31210(图5a:adc,cphhr=3.88,p=0.0008,km时序检验p值=0.00029),gse14814(图5b:adc+scc,cphhr=2.06,p=0.011,km时序检验p值=0.00495),gse13213(图5c:adc,cphhr=2.64,p=0.008,km时序检验p值=0.00586),gse14814(图5d:scc,cphhr=2.9,p=0.032,km时序检验p值=0.02518),gse11969(图5e:adc,cphhr=1.98,p=0.038,km时序检验p值=0.03407)和gse37745(图5f:adc+scc,cphhr=1.41,p=0.042,km时序检验p值=0.04132)的非小细胞肺癌病人有效地分为总体生存期较长和总体生存期较短的亚组。该分类器也可以将多个数据集内的非小细胞肺癌病人分为无复发生存期较长和无复发生存期短的亚组(图6a-cgse8894:adc+scc:cphhr=2.42,p=0.0005,km时序检验p值=0.00032;adc:cphhr=2.29,p=0.02,km时序检验p值=0.01705;scc:cphhr=2.18,p=0.026,km时序检验p值=0.026;图6dgse31210:adc,cphhr=2.07,p=0.005,km时序检验p值=0.00427)。
19-基因表达分类器诊断效能优于其他临床诊断因子
通过一种多因素的cph模型,我们比较了19-基因表达分类器与其他临床诊断因子(如年龄、吸烟历史、基因突变以及基因拷贝数变异等)的诊断效能的优劣。在gse31210中,当19-基因风险因子被纳入到多因素的cph中后,仅有基因突变(kras+或/和egfr+或/和alk+)还能作为独立的诊断因子(p<0.05),而19-基因风险评分仍然是最显著的独立诊断因子(图7a多变量cph,os:hr=3.59,p=0.002;图7brfs:1.95,p=0.013)。值得一提的是在gse13213中,19-基因风险评分甚至超过了肿瘤分期,成为最强的独立诊断因子(图7c多变量cph,os:19-基因风险评分hr=2.65,p=0.01;阶段hr=2.35,p=0.022)。在gse11969中,19-基因风险评分依然是最强的独立诊断因子(图7d多变量cph,os:hr=1.92,p=0.05)
19-基因表达分类器与其他已发表的16-基因分类器的比较
chenetal.在2007年的nejm上发表了一种16-基因表达分类器(17)。我们从文献中获得了16个基因的表达加权系数,因此可以顺利地计算病人的16-基因风险评分。因此可以通过一个双因素的cph模型比较我们的19-基因分类器和该16-基因分类器之间的诊断效能的优劣。结果显示我们的19-基因分类器在包括gse14814(肺腺癌+肺鳞癌)(os:19-基因hr2.14p=0.007;16-基因hr0.63,p=0.09)、gse14814(肺鳞癌)(os:19-基因hr3.05p=0.027;16-基因hr0.75,p=0.536)、gse13213(肺腺癌)(os:19-基因hr2.65p=0.008;16-基因hr1.26,p=0.498)、gse11969(肺腺癌)(os:19-基因hr1.98p=0.007;16-基因hr1.18,p=0.605)、gse37745(肺腺癌+肺鳞癌)(os:19-基因hr1.43p=0.035;16-基因hr1.15,p=0.415)、gse8894(肺腺癌+肺鳞癌)(rfs:19-基因hr2.46p=0.0004;16-基因hr1.57,p=0.067)、gse8894(肺腺癌)(rfs:19-基因hr2.27p=0.022;16-基因hr1.16,p=0.67)和gse8894(肺鳞癌)(rfs:19-基因hr2.04p=0.057;16-基因hr1.19,p=0.633)在内的数据集内都可以更加有效地将非小细胞肺癌病人分为预后良好和预后差的两个亚组。在gse31210(肺腺癌-无复发生存期)模型中,19-基因分类器显示出与16-基因分类器类似的hr(19-基因hr2.05p=0.0059;16-基因hr0.48,p=0.0043)。
应当强调,本发明的上述实施例仅仅是可能的示例实施方式,其仅仅是为了清楚地理解本公开的原理而提出的。在不脱离本公开的精神和原理的情况下,可以对本公开的上述实施例进行许多变化和修改。所有这些修改和变化旨在被包括在本发明的范围内并由所附权利要求保护。
参考文献
1.chenwq,shours,baadepd,etal.cancerstatisticsinchina.2015.cacancerj.clin.2016;66:115-32.
2.burdetts,rydzewskal,tierneyj,etal.postoperativeradiotherapyfornon-smallcelllungcancer.cochranedatabasesystrev2016;9:cd002142.
3.liauwsl,connellpp,weichselbaumrr.newparadigmsandfuturechallengesinradiationoncology:anupdateofbiologicaltargetsandtechnology.scitranslmed2013;5:173sr2.
4.spiottom,fuyx,weichselbaumrr.theintersectionofradiotherapyandimmunotherapy:mechanismsandclinicalimplications.scienceimmunol2016;1:pp.eaag1266.
5.sotiriouc,pusztail.gene-expressionsignaturesinbreastcancer.nengljmed2009;360:790-800.
6.joensuuh.adjuvanttreatmentofgist:patientselectionandtreatmentstrategies.natrevclinioncol2012;9:351-8.
7.ebctcg,petor,daviesc,etal.comparisonsbetweendifferentpolychemotherapyregimensforearlybreastcancer:meta-analysesoflong-termoutcomeamong100,000womenin123randomisedtrials.lancet2012;379:432-44.
8.pusztail.chemotherapyandtherecurrencescore-resultsasexpected?natrevclinioncol2015;12:690-2.
9.paiks,shaks,tangg,etal.amultigeneassaytopredictrecurrenceoftamoxifen-treated,node-negativebreastcancer.nengljmed2004;351:2817-26.
10.sparanoja,grayrj,makowedf,etal.prospectivevalidationofa21-geneexpressionassayinbreastcancer.nengljmed2015;373:2005-14.
11.van‘tveerlj,daih,vandevijvermj,etal.geneexpressionprofilingpredictsclinicaloutcomeofbreastcancer.nature2002;415:530-6.
12.cardosof,van’tveerlj,bogaertsj,etal.70-genesignatureasanaidtotreatmentdecisionsinearly-stagebreastcancer.nengljmed2016;375:717-29.
13.dowsettm,sestaki,lopez-knowlese,etal.comparisonofpam50riskofrecurrencescorewithoncotypedxandihc4forpredictingriskofdistantrecurrenceafterendocrinetherapy.jclinoncol2012;31:2783-90.
14.gnantm,filipitsm,greilr,etal.predictingdistantrecurrenceinreceptor-positivebreastcancerpatientswithlimitedclinicopathologicalrisk:usingthepam50riskofrecurrencescorein1478postmenopausalpatientsoftheabcsg-8trialtreatedwithadjuvantendocrinetherapyalone.annoncol2014;25:339-45.
15.fitzalf,filipitsm,rudasm,etal.thegenomicexpressiontestendopredictisaprognostictoolforidentifyingriskoflocalrecurrenceinpostmenopausalendocrinereceptor-positive,her2neu-negativebreastcancerpatientsrandomisedwithintheprospectiveabcsg8trial.brjcancer2015;112:1405-10.
16.sgroidc,sestaki,cuzickj,etal.predictionoflatedistantrecurrenceinpatientswithoestrogen-receptor-positivebreastcancer:aprospectivecomparisonofthebreast-cancerindex(bci)assay,21-generecurrencescore,andihc4inthetransatacstudypopulation.lancetoncol2013;14:1067-76.
17.chenhy,yusl,chench,etal.afive-genesignatureandclinicaloutcomeinnon-small-celllungcancer.nengljmed2007;356:11-20.
18.chendt,hsuyl,fulpwj,etal.prognosticandpredictivevalueofamalignancy-riskgenesignatureinearly-stagenon-smallcelllungcancer.jnatlcancerinst2011;103:1859-70.
19.luy,lemonw,liupy,etal.ageneexpressionsignaturepredictssurvivalofpatientswithstageinon-smallcelllungcancer.plosmed2006;3:e467.
20.xiey,xiaog,coombeskr,etal.robustgeneexpressionsignaturefromformalin-fixedparaffin-embeddedsamplespredictsprognosisofnon-small-celllungcancerpatients.clincancerres2011;17:5705-14.
21.kratzjr,hej,vandeneedensk,etal.apracticalmolecularassaytopredictsurvivalinresectednon-squamous,non-small-celllungcancer:developmentandinternationalvalidationstudies.lancet2012;379:823-32.
22.director'schallengeconsortiumforthemolecularclassificationoflungadenocarcinoma,sheddenk,taylorjm,etal.geneexpression-basedsurvivalpredictioninlungadenocarcinoma:amulti-site,blindedvalidationstudy.natmed2008;14:822-7.
23.boutrospc,lausk,pintiliem,etal.prognosticgenesignaturesfornon-small-celllungcancer.procnatlacadsciusa2009;106:2824-8.
24.jeongy,xiey,xiaog,etal.nuclearreceptorexpressiondefinesasetofprognosticbiomarkersforlungcancer.plosmed2010;7:e1000378.
25.okayamah,schetteraj,ishigamet,etal.theexpressionoffourgenesasaprognosticclassiferforstageilungadenocarcinomain12independentcohorts.cancerepdemiolbiomarkersprev2014;23:2884-94.
26.zhucq,dingk,strumpfd,etal.prognosticandpredictivegenesignatureforadjuvantchemotherapyinresectednon-small-celllungcancer.jclinoncol2010;28:4417-24.
27.lausk,boutrospc,pintiliem,etal.three-geneprognosticclassifierforearly-stagenon-small-celllungcancer.jclinoncol2007;25:5562-9.
28.roepmanp,jassemj,smitef,etal.animmuneresponseenriched72-geneprognosticprofileforearly-stagenon-small-celllungcancer.clincancerres2009;15:284-90.
29.tangh,xiaog,behrensc,etal.a12-genesetpredictssurvivalbenefitsfromadjuvantchemotherapyinnon-smallcelllungcancerpatients.clincancerres2013;19:1577-86.
30.pardola,stühmerw.therolesofk+channelsincancer.natrevcancer2014;14:39-48.
31.jentschtj.vracsandotherionchannelsandtransportersintheregulationofcellvolumeandbeyond.natrevmolcellbiol2016;17:293-307.
- 还没有人留言评论。精彩留言会获得点赞!