采用定量结构‑活性关系模型预测有机化学品的氯自由基反应速率常数的方法与流程
本发明属于生态风险评价测试策略领域,涉及一种采用定量结构-活性关系模型预测有机化学品的氯自由基反应速率常数的方法。
背景技术:
有机化学品在大气中的氯自由基反应速率常数(kcl)是评估化学品大气持久性的重要参数,对于表征化学品生态环境风险和环境归趋具有重要意义。大气氯自由基(·cl)对有机污染物具有强的化学反应活性,它与大多数有机污染物的反应速率常数比大气中最重要的自由基(羟基自由基)高出一个到多个数量级。但由于过去一直认为·cl主要存在海洋及沿海地带,且其浓度比羟基自由基低(为其浓度的1-10%),导致以前评估的·cl对大气有机污染物转化的贡献低于羟基自由基。然而,氮氧化物与无机氯化物反应这一巨大的·cl内陆来源的突破性发现,使得·cl的大气产生总量得以大幅提升,并且打破了·cl对大气有机污染物转化的地域性限制。因此,·cl在转化大气有机污染物方面起着比以前更为重要的作用,kcl必将成为有机化学品环境风险评价必不可少的参数。
通常,可以通过实验方法如脉冲激光光解-激光诱导荧光(plp-lif)、激光闪光光解-共振荧光(lfp-rf)技术、超低压反应器-四级杆质谱串联法(vlpr-qms)等测得有机物的kcl值。截止目前,仅有五百多种有机化学品具有气相kcl实验数据。然而,人类日常使用的有机化学品已突破14万种,美国化学文摘社(chemicalabstractsservice,cas)登记数据库已包含近一亿三千万种有机和无机化合物,并以每年500~1000种的速度增长,数量十分巨大。如果仅靠实验测定,不仅需要耗费大量的时间、人力和财力,而且时间上存在滞后性,难以满足化学品的“预先防范原则”。因此,有必要发展一种科学、高效、快捷的预测方法。
qsar技术有助于实现有毒有害化学品管理的“预先防范原则”,具有弥补实验数据缺失、克服较高消耗以及减少动物实验等优点,美国、欧洲等发达国家很早就开始运用qsar技术在有毒有害化学品生态风险评价与管理方面进行研究。美国环境保护署(epa)基于qsar技术开发了快速预测软件episuite,已可以预测kow、koc、bcf、生物降解性、空气中的氧化速率(koh,ko3)、水解速率、污水处理厂去除效率等多种环境行为参数。经济合作与发展组织(oecd)于2007年颁布了qsar模型建立与使用的指导文件,提出以下五点原则:(a)包含定义明确的环境指标;(b)采用清晰的数学算法;(c)定义模型的应用域;(d)适当的拟合优度、稳健性及预测能力评价;(e)尽可能的机理解释。
截至目前,已有多个有机化学品kcl预测模型,模型可分为以下五类:(1)对比模型。文献“atmos.environ.,2014,99:159-167.”将logkcl和logkoh相对比,以logkoh为变量建立了logkcl的预测模型。尽管模型具有良好的拟合效果(r2=0.89),但模型只包含了20种不饱和有机化合物,应用域较窄,并且不能解释反应机理。(2)广义拓扑指数模型。文献“j.chem.inf.model.,2007,47(3):794-804.”和“atmos.chem.phys.,2007,7(13):3559-3569.”采用广义拓扑指数作为描述符,建立了有机化学品的气相logkcl预测模型。该类模型虽然在机理解释方面有所提升,但仍然不够清晰,并且应用域较窄。(3)碎片系数模型。文献“chem.phys.lett.,2010,488:135-139.”,“chem.phys.lett.,2009,479:25-29.”,“j.phys.chem.,1992,96,10837-10842.”,“environ.sci.pollut.r.,2015,22(7):4820-4832.”,“int.j.chem.kinet.,2012:52-57.”采用不同的基础公式,针对不同类别的有机化合物分别设定不同的基准系数和取代系数,构建了适用于不同种类有机化合物的小型logkcl预测模型。该类模型虽然在一定程度上反应了取代基对有机化合物logkcl值的影响,机理解释方面有所提升,但仍然不够清晰彻底,并且受公式框架的限制导致应用域较窄。(4)分子轨道模型。文献“atmos.environ.,2014,90:133-145.”,“atmos.environ.,2009,43(38):5996-6002.”和“phys.chem.chem.phys.,2001,3:3981-3986”采用分子轨道描述符ehomo分别构建了适用于不同类别有机化合物的小型logkcl预测模型。该类模型已经可以在一定程度上反映有机化学品的反应机理。但由于描述符数量只有一个且涵盖的化学品数量小,导致模型适用的应用域仍然较窄。(5)其他量子化学模型。文献“chemosphere.2007,67(10):2028-2034.”采用偏最小二乘(pls)分析方法,使用多种易于解释的量子化学描述符构建了包含13种芳香族有机物的气相logkcl预测模型。虽然模型具有良好的拟合效果,而且描述符意义清晰、解释性强。但是模型的应用域同样较窄。文献“j.phys.chem.a.2013,117(30):6433-6449.”采用多元线性回归(mlr)分析方法建立了适用于82种有机化学品的logkcl预测模型。该模型应用域提升较大,并且模型具有一定的解释性。但只适用于单一的h提取反应途径,忽略了cl加成反应途径。
总的来说,目前已有的所有·cl模型都存在应用域狭窄的问题,多数模型机理解释性差,并且没有遵循oecd颁布的qsar模型构建和验证导则。因此,有必要建立一个机理解释清晰、适用有机物种类丰富、反应途径全面的qsar预测模型。并且,依照oecd导则的要求进行模型的验证与应用域的表征。
技术实现要素:
本发明目的是发展一种科学、高效、快捷、应用域广泛、机理解释清晰的有机化学品氯自由基反应速率常数的预测方法。该方法可直接根据有机化学品的分子结构特征预测kcl值,进而预测和评价其环境归趋,为有机化学品的生态风险评价与管理提供必要的基础数据。
本说明书中的“有机化学品”侧重于化学品环境风险评价与管理角度,“有机化合物”侧重于化学品的分子结构角度。
本发明提供一种采用定量结构-活性关系模型预测有机化学品的氯自由基反应速率常数的方法,包括如下步骤,
(1)从大量文献、书籍著作中搜集得到有机化合物的室温(298k)kcl值,如果一种有机化合物含有多个来源的kcl值,则对其取平均值。最终得到温度为298k条件下的506种有机化合物的kcl值。
(2)对于步骤(1)中收集得到的506种有机化合物,使用量子化学计算软件进行电子结构优化。采用b3lyp方法,其中c,o,h,cl,s,f,br,se和n原子,采用6-311+g(d,p)基组;si,i原子,采用lanl2dz基组。使用随机数据分割法将有机化合物以4:1的比例划分为训练集和验证集,训练集有403种有机化合物,验证集有103种有机化合物。训练集中的有机化合物用于构建模型,验证集中的有机化合物用于外部验证。
(3)遗传算法-多元线性逐步回归分析(ga-mlr)模型中描述符的计算:基于步骤(2)优化得到的有机化合物电子结构(对上述506种有机化合物的电子结构进行优化),计算出首次同时使用的、物理意义清晰的量子化学描述65种,其中,基础量子化学描述符17种,四元素描述符18种、符合型量子化学描述符(数学形式变换如(elumo-ehomo)2、(elumo+ehomo)2)21种,并特别加以首次开发使用的量子化学描述符9种进行模型的构建,这9种描述符为qhave,qoave,#nonhatom:c,#x:c,#f:c,#cl:c,#br:c,#i:c,#s:c。与本模型没有采用的、传统上不利于机理解释的dragon描述符相比,本发明的量子化学描述符不仅具有十分清晰的物理意义,同时对有机化合物的kcl具有很好的描述效果,破除了大型qsar模型中描述符物理意义清晰、描述效果好不可兼得的难题。由此,本发明完全使用量子化学描述符所构建的模型,不仅机理解释程度及清晰度得到较大幅度提升,同时实现了很好的拟合效果。
(4)支持向量机-多元线性逐步回归分析(svm-mlr)模型中描述符的计算:基于步骤(2)优化得到的有机化合物电子结构(对上述506种有机化合物的电子结构进行优化),计算描述符1490种,包括dragon描述符1489种,量子化学描述符ehomo1种。
(5)将步骤(1)中收集得到的506种有机化合物的kcl值进行对数变换,获得logkcl形式的数值用于后续模型的构建。
(6)将步骤(3)和(5)中计算得到的物理意义清晰、描述效果好的量子化学描述符和训练集的有机化合物logkcl数值整合,首先使用机器学习方法遗传算法(ga)从65种量子化学描述符中变量选择降维,随后采用多元线性逐步回归分析(mlr)方法去除描述符的多重共线性并构建模型(ga-mlr),模型见式1。将步骤(4)和(5)中计算得到的描述符和训练集的有机化合物logkcl数值整合,使用支持向量机(svm)从1490个描述符中选择出平均影响值(miv)高的前60种描述符,随后采用多元线性逐步回归分析(mlr)方法去除描述符的多重共线性并构建模型(svm-mlr),模型见式2。其中平均影响值miv用于评价各个描述符对于logkcl影响的重要性大小,符号代表相关方向,绝对值大小代表影响的相对重要性。
两种模型的线性关系式如下:
模型一:ga-mlr方法联用的kclqsar模型:
logkcl=3.516+14.33ehomo–0.1380#x:c–8.950qhave–0.8520qcave
-31.17(elumo-ehomo)2–0.5160#nonhatom:c–2.219s–0.6360s-max
+0.0530#h(1)
模型二:svm-mlr方法联用的kclqsar模型。
logkcl=-5.923+15.66ehomo+0.2660ssssch–1.010jgi1+0.3280r3i
–1.137r2e+0.9320r2u+0.5390nr=cp–0.3210hats3u–0.4970nrco
+0.3810dls_05(2)
其中,ehomo表示有机化合物的最高占据分子轨道能量,#x:c表示有机化合物中卤原子数目与碳原子数目的比值,qhave表示氢原子平均电荷,qcave表示碳原子平均电荷,(elumo-ehomo)2表示前沿轨道能级差的平方,#nonhatom:c表示有机化合物中非氢原子数目与碳原子数目的比值,s表示有机化合物的化学软度,s-max表示有机化合物局部软度,#h表示氢原子数目。ssssch与原子过渡态能量有关,jgi1与有机化合物平均拓扑电荷指数有关;r3i、r2e、r2u和hats3u是getawat描述符,r3i与有机化合物电离势有关,r2e与sanderson电负性有关,r2u与lag2状态下的r自相关指数有关,hats3u与lag3状态下的杠杆自相关指数有关;nr=cp表示有机化合物末端sp2杂化的主碳数目,dls_05与改良的药物打分规则有关,nrco表示有机化合物中酮类官能团个数。
所述有机化合物为烷烃、烯烃、炔烃、有机醇、醚、酮、醛、酸、酯、硫代有机物、酰类有机物、有机胺、有机硅、多环芳烃及其取代物、杂环化合物及其衍生物、硝基取代物或卤代有机物。
优选的,所述步骤(2)中进行电子结构优化使用的量子化学计算软件为gaussian09。
优选的,所述步骤(3)中进行量子化学描述符计算所使用的软件为gaussian09或vegazz。
优选的,所述步骤(4)中进行dragon描述符计算所使用的软件为dragon6.0。
本专利还采用去一法与去多法对模型进行内部验证,同时进行了模型的外部验证及评价,使用欧氏距离法对模型进行应用域的表征。模型的验证及评价:模型一的调整决定系数(r2adj)为0.873,均方根误差(rmse)为0.589,表明模型具有良好的拟合优度;去一法交叉验证系数(q2loo)为0.868,bootstrapping验证系数(q2boot)为0.796,表明模型具有良好的稳定性;外部调整决定系数(r2adj.ext)、验证集均方根误差(rmseext)和交叉验证系数(q2ext)分别为0.856、0.593和0.869,表明模型具有良好的预测能力;描述符的变量膨胀因子(vif)均小于10,表明模型不受共线性的影响;r2和q2之差远小于0.3,表明模型不存在过拟合现象。几乎所有数据点的预测误差都在1.5个log单位数值以内,预测效果更好。模型中的数据集数量是其他模型的6倍或更多;不仅同时涵盖原来单个模型所有的有机化合物种类,并且拓展到有机胺、杂环化合物及其衍生物、酰类有机物,极大程度提高了模型的应用域。模型采用了9个物理意义清晰的量子化学描述符,便于推广应用。其中,本发明首次发现:决定有机化合物kcl的主要电子结构特性因素是前沿分子轨道能级差的平方(elumo-ehomo)2,氢原子平均电荷(qhave)和有机化合物软度(s),对于阐明氧化过程中的电子转移机制有重要作用。(elumo-ehomo)2能够比(elumo-ehomo)更加明显的描述电子从elumo激发到ehomo状态所需的能量。qhave从整体上描述有机化合物的反应活性,具体表现为,有机化合物不同的碳框架及其上取代的不同官能团,会通过空间位阻、极性效应等多种影响因素影响有机化合物稳定的几何构型,进而影响其在氢提取反映过程中得失电子的能力,qhave对这些影响因素进行综合性的描述。s描述有机化合物的极性效应。模型二调整决定系数(r2adj)为0.836,均方根误差(rmse)为0.671,表明模型具有较好的拟合优度;去一法交叉验证系数(q2loo)为0.828,bootstrapping验证系数(q2boot)为0.794,表明模型具有较好的稳定性;外部调整决定系数(r2adj.ext)、验证集均方根误差(rmseext)和交叉验证系数(q2ext)分别为0.853、0.613和0.852,表明模型具有较好的预测能力;描述符的变量膨胀因子(vif)均小于10,表明模型不受共线性的影响;r2和q2之差远小于0.3,表明模型不存在过拟合现象。两个qsar预测中相比,模型一的预测效果更好,模型二的降维效果更强。
使用欧氏距离法对模型进行应用域的表征:欧氏距离定义为有机物的特征向量到描述符空间中心的特征向量的距离,公式见(3)。采用ambitdiscovery软件计算有机化学品的欧氏距离(di),并将di在训练集中的最大值作为警戒值。
其中,xik与xk分别为第k个描述符值与描述符平均值。模型一训练集中的欧几里得距离最大值为d*=1.47,模型二训练集中的欧几里得距离最大值为d*=1.49,两个模型所有数据点的欧几里得距离均在警戒值范围之内,同时绝大部分数据点的预测误差在范围之内。结果表明,本发明的两个模型适用于上述提及的有机化合物。
本发明的有益效果是,采用本发明可以通过分子结构特征科学、快速地预测有机化学品的logkcl值。该方法不仅适用的应用域广泛,机理解释性强,而且简单快捷、成本低廉,节省了实验测定所需的人力、物力和财力。本发明涉及的logkcl预测方法的建立和验证严格依据oecd颁布的qsar模型建立与使用导则,可为有机化学品监管提供数据支持。有益效果具体表现为以下几点:
1.模型一使用非传统的机器学习方法ga进行变量选择以降维,随后联用透明度高的mlr方法去除多重共线性并建模(ga-mlr);首次完全采用65个物理意义清晰的量子化学描述符构建模型,尤其部分描述符为创新性的首次开发使用。与传统的几乎全由dragon描述符组成的模型、单纯采用非透明算法建模的qsar模型相比,本专利不仅能选择出重要性高的描述符以减少变量数量,并且模型在反应机理的可解释程度以及透明度方面获得较大幅度的提升。
2.模型一绝大部分数据集点的logkcl预测误差都在1.5个log单位值以内,模型的预测效果更好。加之上一点中对于反应机理可解释程度及模型透明度的提升,本专利破除了大型qsar模型中描述符物理意义清晰、描述效果好不可兼得的难题。
3.本发明从模型一中首次发现:决定有机化合物kcl的主要电子结构特性因素是前沿分子轨道能级差的平方(elumo-ehomo)2,氢原子平均电荷(qhave)和有机化合物软度(s)。同时加入了前人·cl模型所缺少的机理解释,对于阐明氧化反应的电子转移机制具有重要作用。
4.模型二在建立过程中,先将1890个描述符筛选至60个再构建模型,具有很强的降维能力。
5.模型中的有机化合物数目,是前人模型的6倍或更多;不仅同时涵盖原来单个模型所有的有机化合物种类,并且拓展到有机胺、杂环化合物及其衍生物、酰类有机物,极大程度提高了模型的应用域。本模型适用的有机化合物种类有烷烃、烯烃、炔烃、有机醇、醚、酮、醛、酸、酯、硫代有机物、酰类有机物、有机胺、有机硅、多环芳烃及其取代物、杂环化合物及其衍生物、硝基取代物或卤代有机物。
6.本发明为首个严格遵循oecd颁布的qsar模型构建与验证导则的kcl预测模型,具有良好的拟合优度、稳健性和预测能力,更加完善的契合了导则中“尽可能的机理解释”原则。模型简约,便于推广应用,可为生态风险性评价和监管提供数据支持;
7.与前人的涵盖有机化合物数目最多的模型相比,本发明适用于预测包含h提取、cl加成反应途径在内的全局反应速率常数,更贴近化学品环境风险评价的实际需求。
附图说明
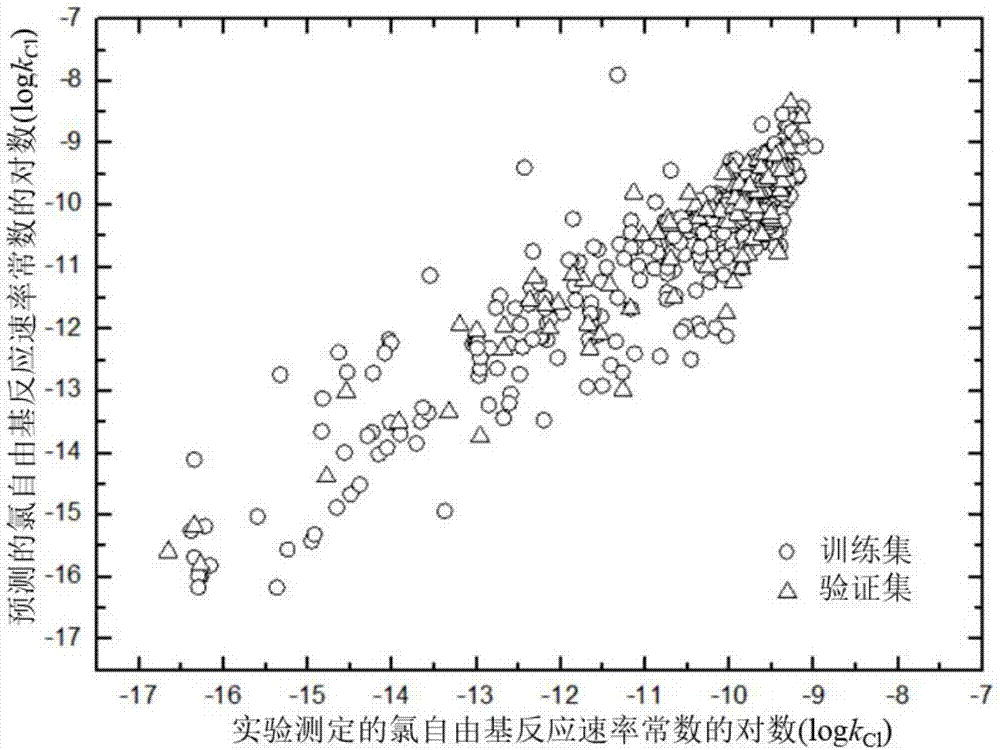
图1为ga-mlr模型(式1)训练集和验证集的logkcl的实测值与预测值的拟合图,训练集化合物为403种,验证集化合物为103种。
图2为svm-mlr模型(式2)训练集和验证集的logkcl的实测值与预测值的拟合图,训练集化合物为403种,验证集化合物为103种。
图3为ga-mlr模型基于欧几里得距离法的应用域表征图。
图4为svm-mlr模型基于欧几里得距离法的应用域表征图。
图5为有ga-mlr模型的有机化合物标准残差对欧几里得距离表征图。
图6为有svm-mlr模型的有机化合物标准残差对欧几里得距离表征图。
具体实施方式
下面结合说明书附图和实施例,对本发明的具体实施方式作进一步详细描述。以下实施例仅用于说明本发明,但不能用来限制本发明的范围。
实施例1
预测有机化学品的氯自由基反应速率常数的方法,包括如下步骤:
首先,数据收集:从大量文献、书籍著作中搜集得到有机化合物的室温(298k)kcl值,如果一种有机化合物含有多个来源的kcl值,则对其取平均值。最终得到温度为298k的506种有机化合物的kcl值,有机化合物为烷烃、烯烃、炔烃、有机醇、醚、酮、醛、酸、酯、硫代有机物、酰类有机物、有机胺、有机硅、多环芳烃及其取代物、杂环化合物及其衍生物、硝基取代物或卤代有机物。
其次,电子结构优化、数据集分组:在量子化学计算软件gaussian09中对数据集中所有的有机化学品进行了结构优化,采用b3lyp方法。其中c,o,h,cl,s,f,br,se和n原子,采用6-311+g(d,p)基组;si,i,pb和hg原子,采用lanl2dz基组。采用随机数据分割法将有机物以4:1的比例划分为训练集和验证集,训练集有403种有机化合物,验证集有103种有机化合物。
再次,分子结构描述符的计算、对数形式变换和模型的建立:(1)ga-mlr模型构建所使用的分子结构描述符均为物理意义清晰、有利于机理解释的量子化学描述符。本实施例引入基础量子化学描述符17种,四元素描述符18种、符合型量子化学描述符21种,并特别加以首次使用的量子化学描述符9种用于进行模型的构建,这9种描述符为qhave,qoave,#nonhatom:c,#x:c,#f:c,#cl:c,#br:c,#i:c,#s:c。描述符计算所使用的软件有gaussian09,vegazz等。svm-mlr模型构建所使用的描述符共计1490种,其中dragon描述符1849种,量子化学描述符1种(ehomo),所需补充使用的软件为dragon6.0。(2)对有机化合物kcl值进行对数形式上的变换。(3)使用上述物理意义清晰的65个量子化学描述符和训练集中的logkcl值,先采用机器学习方法遗传算法(ga)对量子化学描述符进行变量选择以降维,再联用多元线性逐步回归分析(mlr)方法去除描述符的多重共线性并构建模型,得到的ga-mlr模型见式1。使用上述1490种描述符和训练集的有机化合物logkcl数值,先采用支持向量机(svm)从1490个描述符中选择出平均影响值(miv)高的前60种描述符,随后采用mlr方法去除描述符的多重共线性并构建模型(svm-mlr),模型见式2。其中平均影响值miv用于评价各个描述符对于logkcl影响的重要性大小,符号代表相关方向,绝对值大小代表影响的相对重要性。
logkcl=3.516+14.33ehomo–0.1380#x:c–8.950qhave–0.8520qcave
-31.17(elumo-ehomo)2–0.5160#nonhatom:c–2.219s–0.6360s-max
+0.0530#h(1)
logkcl=-5.923+15.66ehomo+0.2660ssssch–1.010jgi1+0.3280r3i
–1.137r2e+0.9320r2u+0.5390nr=cp–0.3210hats3u–0.4970nrco
+0.3810dls_05(2)
其中,ehomo表示有机化合物的最高占据分子轨道能量,#x:c表示有机化合物中卤原子数目与碳原子数目的比值,qhave表示氢原子平均电荷,qcave表示碳原子平均电荷,(elumo-ehomo)2表示前沿轨道能级差的平方,#nonhatom:c表示有机化合物中非氢原子数目与碳原子数目的比值,s表示有机化合物的化学软度,s-max表示有机化合物局部软度,#h表示氢原子数目。ssssch与原子过渡态能量有关,jgi1与有机化学品平均拓扑电荷指数有关;r3i、r2e、r2u和hats3u是getawat描述符,r3i与有机化合物电离势有关,r2e与sanderson电负性有关,r2u与lag2状态下的r自相关指数有关,hats3u与lag3状态下的杠杆自相关指数有关;nr=cp表示有机化合物末端sp2杂化的主碳数目,dls_05与改良的药物打分规则有关,nrco表示有机化合物中酮类官能团个数。
最后,模型的验证与评价、应用域表征:进行模型的拟合优度、稳健性及预测能力验证,对模型进行了评价,并使用欧氏距离法进行应用域的表征(见图1-图6)。由模型一训练集的内部验证参数r2adj(0.873),rmse(0.589),q2loo(0.868),q2boot(0.796),可知模型具有良好的拟合优度和稳健性;由验证集获得的外部验证参数r2adj.ext(0.856),rmseext(0.593),q2ext(0.869),可知模型具有良好的预测能力。由描述符的vif小于10可知模型不具有共线性,由r2和q2之差远小于0.3可知模型不存在过拟合现象。模型中的数据集数量可知本发明极大程度提高了模型的应用域。模型中采用了9个物理意义清晰的量子化学描述符,便于推广应用,其中,本发明首次发现:决定有机化合物kcl的主要电子结构特性因素是前沿分子轨道能级差的平方(elumo-ehomo)2,氢原子平均电荷(qhave)和有机化合物软度(s),并对以上三个描述符进行了机理解释。由模型二训练集的内部验证参数r2adj(0.836),rmse(0.671),q2loo(0.828),q2boot(0.794),可知模型具有较好的拟合优度和稳健性;由验证集获得的外部验证参数r2adj.ext(0.853),rmseext(0.613),q2ext(0.852),可知模型具有较好的预测能力。由描述符的vif小于10可知模型不具有共线性,由r2和q2之差远小于0.3可知模型不存在过拟合现象。两个qsar预测模型相比较,模型一的预测效果更好,模型二的降维效果更强。此外,还使用欧氏距离法对两个模型进行了应用域的表征。
实施例2
给定一个有机物6-硝基间甲酚,要预测其logkcl值。首先基于6-硝基间甲酚的结构信息,使用gaussian09软件包进行电子结构优化。然后基于以上结果计算出ehomo,#x:c,qhave,qcave,(elumo-ehomo)2,#nonhatom:c,s,s-max,#h值分别为-0.261,0,0.168,-0.091,0.022,1.571,3.412,0.562,7。使用ambitdiscovery软件计算有机化学品的di值为0.486(<1.472),因此该有机物在应用域范围内。将上述描述符值带入实施例1的公式(1),得到logkcl的预测值为-10.716,其实验测定值为-10.712,残差值0.0037,预测值和实验值符合较好。
实施例3
给定一个有机物巴豆醛,要预测其logkcl值。首先基于巴豆醛的结构信息,使用gaussian09软件包进行电子结构优化。然后基于以上结果计算出ehomo,#x:c,qhave,qcave,(elumo-ehomo)2,#nonhatom:c,s,s-max,#h值分别为-0.264,0,0.142,-0.139,0.038,1.25,2.743,1.184,6。使用ambitdiscovery软件计算有机化学品的di值为0.388(<1.472),因此该有机物在应用域范围内。将上述描述符值带入实施例1的公式(1),得到logkcl的预测值为-9.753,其实验测定值为-9.569,残差值0.1837,预测值和实验值符合较好。
实施例4
给定一个有机物二甲基亚砜,要预测其logkcl值。首先基于二甲基亚砜的结构信息,使用gaussian09软件包进行电子结构优化。然后基于以上结果计算出ehomo,#x:c,qhave,qcave,(elumo-ehomo)2,#nonhatom:c,s,s-max,#h值分别为-0.239,0,0.180,-0.573,0.054,2,2.756,1.249,6。使用ambitdiscovery软件计算有机化学品的di值为0.504(<1.472),因此该有机物在应用域范围内。将上述描述符值带入实施例1的公式(1),得到logkcl的预测值为-10.336,其实验测定值为-10.148,残差值0.1878,预测值和实验值符合较好。
实施例5
给定一个有机物萘己环,要预测其logkcl值。首先基于萘己环的结构信息,使用gaussian09软件包进行电子结构优化。然后基于以上结果计算出ehomo,#x:c,qhave,qcave,(elumo-ehomo)2,#nonhatom:c,s,s-max,#h值分别为-0.213,0,0.142,-0.118,0.029,1,3.412,0.534,10。使用ambitdiscovery软件计算有机化学品的di值为0.614(<1.472),因此该有机物在应用域范围内。将上述描述符值带入实施例1的公式(1),得到logkcl的预测值为-9.495,其实验测定值为-9.521,残差值-0.026,预测值和实验值符合较好。
实施例6
给定一个有机物1,2,2-三氟乙基三氟甲基醚,要预测其logkcl值。首先基于1,2,2-三氟乙基三氟甲基醚的结构信息,使用gaussian09软件包进行电子结构优化。然后基于以上结果计算出ehomo,#x:c,qhave,qcave,(elumo-ehomo)2,#nonhatom:c,s,s-max,#h值分别为-0.377,2,0.202,0.206,0.145,3.333,1.954,0.335,2。使用ambitdiscovery软件计算有机化学品的di值为0.987(<1.472),因此该有机物在应用域范围内。将上述描述符值带入实施例1的公式(1),得到logkcl的预测值为-14.831,其实验测定值为-14.921,残差值-0.0903,预测值和实验值符合较好。
实施例7
给定一个有机物丙酸丁酯,要预测其logkcl值。首先基于丙酸丁酯的结构信息,使用gaussian09软件包进行电子结构优化。然后基于以上结果计算出ehomo,ssssch,jgi1,r3i,r2e,r2u,nr=cp,hats3u,nrco,dls_05值分别为-0.281,0.000,0.188,2.68,1.837,1.962,0.000,0.804,0.000,1.000。使用ambitdiscovery软件计算有机化学品的di值为0.418(<1.485),因此该有机物在应用域范围内。将上述描述符值带入实施例1的公式(2),得到logkcl的预测值为-9.766,其实验测定值为-9.801,残差值-0.035,预测值和实验值符合较好。
实施例8
给定一个有机物顺式-4-甲基-2-戊烯,要预测其logkcl值。首先基于顺式-4-甲基-2-戊烯的结构信息,使用gaussian09软件包进行电子结构优化。然后基于以上结果计算出ehomo,ssssch,jgi1,r3i,r2e,r2u,nr=cp,hats3u,nrco,dls_05的值分别为-0.247,0.718,0.300,2.160,1.736,1.897,0.000,0.732,0.000,0.500。使用ambitdiscovery软件计算有机化学品的di值为0.306(<1.485),因此该有机物在应用域范围内。将上述描述符值带入实施例1的公式(2),得到logkcl的预测值为-9.443,其实验测定值为-9.386,残差值0.057,预测值和实验值符合较好。
本发明实施例2-8中,所选取的有机化合物的官能团各不相同,涉及不同的有机化合物类别。实施例展示出模型广泛的应用域和较好的拟合效果。
- 还没有人留言评论。精彩留言会获得点赞!