一种筛选慢性萎缩性胃炎大鼠生物标志物的装置的制作方法
本发明涉及生物标志物筛选领域,特别涉及慢性萎缩性胃炎大鼠生物标志物筛选领域。
背景技术
代谢组学作为一门新兴的系统生物学技术,主要用于研究生物体受到外界环境、疾病、药物等干预后其代谢产物种类和数量的变化及变化规律。其以高通量检测和数据处理为手段,以组群指标分析为基础,能够描述病症的整个代谢过程所产生内源性代谢产物的动态变化,被广泛用于药物的作用机制和靶点研究中。代谢组学是一种相对较新的“组学”技术,试图描述所有低分子量代谢物,常用于纵向研究,如监测疾病进展,追踪营养干预或观察药物毒性。
代谢组学数据最大特点是变量数远远大于观测数,且变量之间存在着高度相关性。目前,最常用于代谢组学数据分析的方法有:无监督学习方法的主成分分析(pca)、有监督学习方法的偏最小二乘判别分析(pls-da)、正交偏最小二乘判别分析(opls-da)等。这些方法可以提取原始信息的最大变异或在此基础上的最佳解释变异,将高维数据映射到低维空间,并给出降维后数据的可视化展示。然而,这些传统的静态分析方法多从病理状态的单一横断面入手研究,未充分考虑疾病病理过程的复杂性,因而易得到相对片面的结论,不能全面反映生理病理演变规律。
随着研究深入,代谢组学不再拘泥于横断面研究,包含时间因素的动态代谢组数据被越来越多的研究所获得,这使得传统分析方法不再适用。因此,在数据分析过程中需要考虑其他因素以及感兴趣的生物状况。基于方差同步主成分分析(anova-simultaneouscomponentanalysis,asca)专门设计的统计方法已经被开发来处理多因素问题以及时间序列研究。它已成功应用于分析代谢组学数据。
把数据集分解成效应矩阵同时进行降维分析。以基于实验设计、考虑时间因素的代谢数据为例,asca首先将总变异分解成三个来源:时间因素,时间因素与处理因素的交互效应,以及不同个体间的变异。分别对这三部分矩阵进行主成分分析,得到随着时间的改变各部分效应的得分变化趋势图,从而给出合理的生物学解释。
慢性萎缩性胃炎(chronicatrophicgastritis,cag)是消化系统的常见疾病之一,是指胃黏膜上皮遭受反复损害导致固有腺体的减少,伴或不伴纤维替代,肠腺化生和/或假幽门腺化生的一种慢性胃部疾病。可以利用代谢组学方法筛选潜在生物标志物,用于临床和药物研究中,探究发病机制等。
技术实现要素:
本发明所要解决的问题是:如何提供一种可靠的萎缩性胃炎模型生物标志物的评价筛选装置。
本发明所采用的技术方案是:一种筛选慢性萎缩性胃炎大鼠生物标志物的装置,包括箱体、安装在箱体上的频谱仪、安装在箱体上的数据处理芯片、安装在箱体上的显示器,将经过预处理的尿样装入频谱仪的核磁管,开启频谱仪采集1dnmr数据,并将该数据作为原始数据输入数据处理芯片,数据处理芯片对原始数据首先进行预处理,采用simca-p软件对预处理后的数据进行多元统计分析,通过pls-da分析获得时间依赖性尿液动态代谢组学特征,然后使用asca模型分析cag表型、时间及其相互作用对尿样中代谢变化的影响,鉴定与之作用相关的代谢物,最后通过显示器显示出来。
作为一种优选方式:尿样的预处理过程为:在500μl的尿样中加入200μl磷酸缓冲液,混匀后离心,取上清550μl作为预处理的尿样,磷酸缓冲液按照质量百分比含10%d2o和0.05%tsp,ph=7.4。
作为一种优选方式:频谱仪采用brukeraviii600,对应的质子共振频率600.13mhz,采集时温度控制为298k,采用bbo探头,测样前对频谱仪进行温度校正,匀场校正。
作为一种优选方式:数据处理芯片对原始数据进行预处理的过程为,通过mestrenova对所有的原始数据进行相位校正和基线校正,尿样的1d和2dnmr谱参考tsp的化学位移,以δ0.001ppm积分分段对1dδ0.50-9.00ppm区域进行积分,且排除δ4.68-5.19ppm区域以消除不完全的含水饱和度的影响,然后将生成的数据修改为metaboanalyst分析的'时间序列/双因子设计'模块中描述的可接受数据格式,该模块包含实验动物中多种类型的变异、时间的变化及其相互作用,数据集归一化为常量和paretoscaling预处理。
作为一种优选方式:采用simca-p软件对预处理后的数据进行多元统计分析,通过pls-da分析获得时间依赖性尿液动态代谢组学特征的具体过程为,将预处理的数据采用simca-p软件进行多元统计分析,将数据引入pls-da分析,观察代谢差异,由于时间的推移,显现出主要的数据扰动源是由大鼠的生长所代表的,轨迹显示对照组在可控范围内移动,模型组从0周到第10周的代谢轮廓随时间而变化。
作为一种优选方式:使用asca模型分析cag表型、时间及其相互作用对尿样中代谢变化的影响,鉴定与之作用相关的代谢物,最后通过显示器显示出来的具体过程为,用asca模型将尿动态代谢组学数据分成不同的子集,描述动物实验之间的差异,时间变化以及它们之间的相互作用,通过置换方法进行显着性检验,使用显示特征值与因子之间关系的图像,与因子a、因子b及其相互作用相关的主要模式显示在基于相应子模型的pc1的得分散点图中,使用基于杠杆效率(leverage)/平方预测误差(squaredpredictionerror,spe)图确定与特定因子相关的重要变量,显示根据表型,时间及其相互作用的因素,通过asca模型鉴定的代谢物有:丙氨酸、脂质、肌酸、二甲基甘氨酸、α-酮戊二酸、丙氨酸、琥珀酸,α-酮戊二酸、丙氨酸、马尿酸和尿囊素作为候选生物标志物,最后将检测到的候选生物标志物在显示器上显示出来。
本发明的有益效果是:本发明更全面灵敏,综合地体现代谢过程中标志物筛选的合理性和科学性,提供一种可靠的萎缩性胃炎模型生物标志物的评价筛选装置。
附图说明
图1模型复制后不同采集时间下偏最小二乘法-判别分析(pls-da)观察尿代谢差异变化;
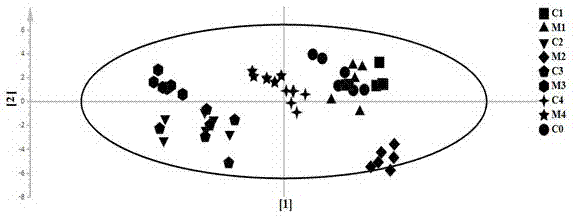
图2表型因子(对照和模型)相关的主要模式的得分散点图;
图3时间变化相关的主要模式的得分散点图;
图4时间变化与表型因子(对照和模型)相互作用相关的主要模式的得分散点图;
图5cag大鼠尿液样本opls-da分析得分图及载荷图。
图2中坐标轴【1】和【2】分别为第一主成分和第二主成分。c:对照组,m:模型组;c0:0周采集的尿样,c1和m1:4周采集尿样,c2和m2:6周采集的尿样,c3好和m3:8周的采集尿样,c4和m4:10采集的尿样
具体实施方式
实施例1
1.大鼠慢性萎缩性胃炎模型复制
sd大鼠,雄性,体重180~220g,购自维通利华实验动物有限公司,许可证:scxk(京)2014-2015。动物共分为2组,每组6只:正常对照组(controlgroup):不给予刺激,给予生理盐水,普通饲料喂养;病理模型组(caggroup):①脱氧胆酸钠自由饮用:浓度为20mmo1/l的脱氧胆酸钠溶液从造模的第一天开始,每单日自由饮用,保证足量;②氨水自由饮用:浓度为0.1%的氨水溶液从造模第一天开始,每双日自由饮用,保证足量;③饥饱失常法:两天足食,1天禁食。循环实施。
2.尿液样本收集及预处理
禁食12小时后,在代谢笼中收集第0、4、6、8和10周禁食12h尿液(包含0.05%叠氮钠)。全尿样品在3,333g下离心10min。然后将上清液小心收集在新鲜的聚丙烯管中,储存在-80℃下进一步分析。
取上述收集的大鼠尿样500μl,加200μl磷酸缓冲液(含10%d2o和0.05%tsp,ph=7.4),混匀后离心,取上清550μl加入本发明的核磁管中待测。
3.nmr测试条件
所有nmr数据在brukeraviii600型谱仪上采集,对应的质子共振频率600.13mhz,实验温度298k,bbo探头。测样前对谱仪进行温度校正,匀场校正。采用以下脉冲序列进行分析:noesygppr1d脉冲序列(recycledelay(rd)-90°_-t1-90°_-tm-90°_-acq),水峰压制相关脉冲序列。选取参数如下:90°脉冲长度为10μs,谱宽12345.7hz,混合时间tm100ms,t13μs,驰豫时间(recycledelay)1.0s,采样点数(td)为96k,信号累加次数(ns)为64。
4.数据预处理
通过mestrenova(version8.0.1,mestrelabresearch,santiagodecompostella,spain)。对所有的1hnmr原始数据进行相位校正和基线校正。尿液的1d和2dnmr谱参考tsp(δ0.00ppm)的化学位移。以δ0.001ppm积分分段对1dδ0.50-9.00ppm区域进行积分,且排除δ4.68-5.19ppm区域以消除不完全的含水饱和度的影响。
然后将生成的数据修改为metaboanalyst分析的'时间序列/双因子设计'模块中描述的可接受数据格式,该模块包含实验动物(模型)中多种类型的变异、时间的变化及其相互作用。数据集归一化为常量和paretoscaling预处理。
5.数据的多元统计分析
将预处理的数据采用simca-p软件(umetricsab,sweden,13.0version)进行多元统计分析,将数据引入pca分析,使用偏最小二乘法-判别分析(pls-da),观察代谢差异(图1),由于时间的推移,显现出主要的数据扰动源是由大鼠的生长所代表的。轨迹显示对照组在可控范围内移动。模型组从0周位置沿pc2轴移动到6周后,然后沿着pc3轴移动到8周,并在第10周结束时达到最大位移。结果表明,代谢变化是随时间而变化的。为了便于数据探索和模式发现,开展了两种可视化方法双向热图以揭示潜在生物标志物的动态改变。
asca模型可以将总变异分成对应于实验因子的贡献。在这里,asca被用来将尿动态代谢组学数据分成不同的子集,描述动物实验之间的差异,时间变化以及它们之间的相互作用。在后续过程中,通过置换方法进行显着性检验以验证模型,如表型、时间水平及其相互作用,显着性水平用p<0.05证明。对于包含多种类型变化的多级数据集,作为确定要提取因子数量的第一阶段的一部分,使用了一个显示特征值与因子之间关系的图像。与因子a(时间变化)、因子b(表型,对照和模型)及其相互作用相关的主要模式显示在基于相应子模型的pc1的得分散点图(图2)中。基于杠杆效率(leverage)/平方预测误差(squaredpredictionerror,spe)图(图2-4)确定与特定因子相关的重要变量。显示根据表型,时间及其相互作用的因素,通过asca模型鉴定的代谢物有:丙氨酸、脂质、肌酸、二甲基甘氨酸、α-酮戊二酸、丙氨酸、琥珀酸,α-酮戊二酸、丙氨酸、马尿酸和尿囊素作为候选生物标志物。
表1基于asca模型鉴定的与表型、时间及其相互作用相关的代谢物
实施例2
1.大鼠慢性萎缩性胃炎模型复制
sd大鼠,雄性,体重180~220g,购自维通利华实验动物有限公司,许可证:scxk(京)2014-2015。动物共分为2组,每组6只:正常对照组(controlgroup):不给予刺激,给予生理盐水,普通饲料喂养;病理模型组(caggroup):①脱氧胆酸钠自由饮用:浓度为20mmo1/l的脱氧胆酸钠溶液从造模的第一天开始,每单日自由饮用,保证足量;②氨水自由饮用:浓度为0.1%的氨水溶液从造模第一天开始,每双日自由饮用,保证足量;③饥饱失常法:两天足食,1天禁食。循环实施。
2.尿液样本收集及预处理
禁食12小时后,在代谢笼中收集第0、4、6、8和10周禁食12h尿液(包含0.05%叠氮钠)。全尿样品在3,333g下离心10min。然后将上清液小心收集在新鲜的聚丙烯管中,储存在-80℃下进一步分析。
取上述收集的大鼠尿样500μl,加200μl磷酸缓冲液(含10%d2o和0.05%tsp,ph=7.4),混匀后离心,取上清550μl加入本发明装置的核磁管中待测。
3.nmr测试条件
所有nmr数据在brukeraviii600型谱仪上采集,对应的质子共振频率600.13mhz,实验温度300k,bbi探头。测样前对谱仪进行温度校正,匀场校正。采用以下脉冲序列进行分析:noesygppr1d脉冲序列(recycledelay(rd)-90°-t1-90°-tm-90°-acq),水峰压制相关脉冲序列。选取参数如下:90°脉冲长度为10μs,谱宽20ppm,混合时间tm100ms,t13μs,驰豫时间(recycledelay)2s,采样点数(td)为96k,信号累加次数(ns)为64。
4.数据处理
通过mestrenova(version8.0.1,mestrelabresearch,santiagodecompostella,spain)。对所有的1hnmr原始数据进行相位校正和基线校正。尿液的1d和2dnmr谱参考tsp(δ0.00ppm)的化学位移。以δ0.001ppm积分分段对1dδ0.50-9.00ppm区域进行积分,且排除δ4.68-5.19ppm区域以消除不完全的含水饱和度的影响。将分段积分数据按图谱总面积归一化处理后生成数据矩阵,导入多元统计分析软件进行统计分析。
5.数据的多元统计分析
将预处理的数据采用simca-p软件(umetricsab,sweden,13.0version)进行多元统计分析,应用正交偏最小二乘判别分析(opls-da)对数据进行多元统计分析。通过载荷图对变量加载的结果进行描述,结合变量重要性(vip)分析,从对照组与模型组中找到含量变化差异显著的变量,利用其相应s-plot图结合vip值来鉴定潜在生物标志物,s-plot图中离原点越远的点及vip>1的化合物定为潜在生物标志物(见图3)。组间差异代谢产物的信息及在各组含量变化趋势见表2。
表2.尿液中cag相关的潜在生物标志物
与正常对照组相比,*p<0.05,**p<0.01。
本实施例也对比例相比较,具有高效、快速、无创伤、特异性强的优点,实施例利用方差同步主成分分析用于动态代谢组学数据的分析,分析模型表型、时间及其相互作用对大鼠尿液中代谢变化的影响。找到可以,通过分析造模前后机体终端产物尿液中的18个生物标记物积分均值的变化针对性地评价慢性萎缩性胃炎的模型。
- 还没有人留言评论。精彩留言会获得点赞!