一种获得基因单体型序列的方法及其应用与流程
本发明属于生物信息学领域,具体涉及一种获得基因单体型序列的方法及其应用。
背景技术
目前获得单体型的方法,主要有家系分型、群体连锁不平衡分型、物理分型。
家系分型的过程,直观、简单、准确,不需要进行复杂的统计学计算。根据父母的特异位点,确定子代的杂合位点两种等位型哪个源于父亲,哪个源于母亲,是筛查母源性或父源性疾病的重要方法。但相比于只测子代的方法,家系分析分析需要额外测其亲本,增加了较多的成本。
群体连锁不平衡分型方法,是利用群体中大量无血缘关系的个体,依据基本的连锁不平衡遗传原理和相关模型,推断群体中每个个体单倍型的方法。该方法需要大量的计算,同时其精度受群体的制约,且不适用于个体特异性较强的变异。
物理定相、分型,由于每条下机数据必定来源于一个染色体,对于每一个测序片段而言,其本身就是一个单倍体的“局部”;二代测序片段较短,需要依赖较多的杂合位点进行拼接,从而接出完整的单体型。而三代测序序列足够长,在足够的深度下,可以有效排除测序错误的影响,达到个体的分相,从而获得准确的单体型。
目前可用于三代测序的获得基因单体型序列的方法有maginphase法和laa法。
maginphase法经过samtools初步得到snp,在此基础上再由whatshap采用最小路径法对snp进行粗分相,然后在此基础上maginphase采用隐马尔科夫模型,进一步分相。然而,maginphase法有如下缺陷:
1、对于多态性特别高的区域不敏感;
2、单体型不精准,某些多态性位点易推断错误;
3、maginphase对深度有要求,只能承受15x左右的计算量,对于错误率较高的情况,不能有效利用深度消除测序错误,降低了结果的准确性。
laa(longampliconanalysis软件)经过聚类,每个扩增子聚为一类(cluster),然后用pagerank算法对cluster的序列(subreads)进行排序;采用arrow算法检测snp和产生一致性序列。laa法的缺陷是对于多态性特别高的序列,产生单体型易存在错误snp。
技术实现要素:
为了解决上述技术问题,本发明的一方面提供了一种获得基因单体型序列的方法,所述方法包括步骤:
1)生成矩阵
将基因测序原始数据进行ccs(circularconsensussequencing)矫正,将得到的ccs序列与参考序列进行长序列比对,输出比对矩阵m,矫正纯合位点的深度小于5%的碱基,重新调整矩阵,形成粗矫正的矩阵m1;
2)分相,包括步骤:
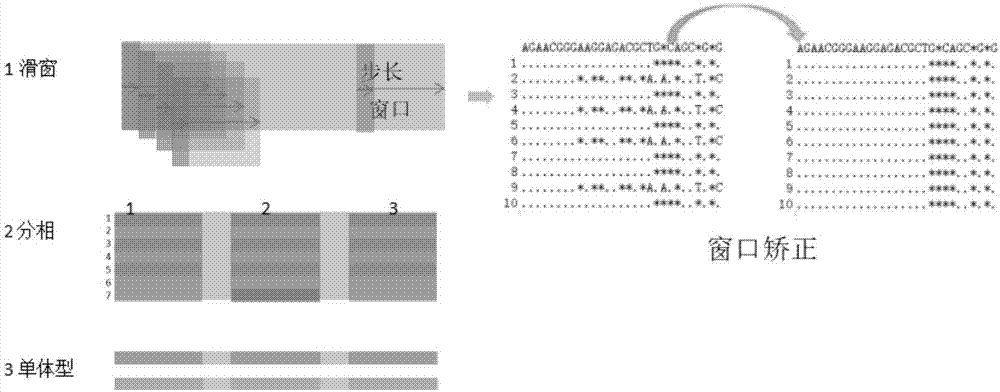
2.1)按窗口编码归一化矩阵
编码m1,设置窗口长度与步长,每个窗口对应的矩阵横坐标为i、纵坐标为j,用w[i]表示该窗口,w[i,j]表示窗口中的每条序列对应的矩阵元素,窗口长度为win,步长为step;
窗口每滑动一步,调整当前窗口的矩阵元素,调整规则以w[i]内序列相同的行,转换为同一种比对矩阵,w[i]则形成一个新的矩阵m[i];
当前步长内的元素被确定,m[i]步长外的元素将继续进入下一个窗口继续调整;
整个扩增子经过一步一个窗口滑动,最终形成按照“步长和窗口”编码新的矩阵m2;
2.2)筛选杂合分相窗口
对于矩阵m2,其每一步的窗口w[i],若窗口内每条矩阵元素w[i,j]对应的同种序列类型深度之和,只有一种超过了总深度的5%,则取消该窗口参与分相的资格;
否则若有大于5%的两种或以上序列类型,则标记为用来分相的窗口w[i]∈h;下一个被标记的窗口,和该窗口无交集;若同时相邻的窗口被标记为h,则所有的这些窗口合并为一个新的窗口w_combine[i]将重新作为一个窗口进行调整,即w_combine[i]内序列相同的行,转换为同一种比对矩阵,形成一个新的矩阵m_combine[i];
被标记的杂合窗口形成了新的杂合矩阵h;
3)相位的确定
假设杂合矩阵h由n个被编码的窗口,矩阵的深度为d;
随后进行k轮循环分相:
随机选择k个小于d的数字,假设第t次随机选择了r,t∈random{1..k},r∈random{0..d},每个窗口的纵坐标为r的行,作为参考;每个窗口内均有两种最大深度的矩阵类型w[r,max,]、w[r,second,],这两种类型哪一种和参考的相似度高,将暂时归为一类phase_temp[1],另一种归为另一类phase_temp[2];窗口内每行根据与w[r,max,]、w[r,second,]的相似度划分类;
窗口内某行与phase_temp[1]、phase_temp[2]相似度等于相同碱基除以窗口长度,对于每一行计算最终属于phase_temp[1]、还是phase_temp[2]的得分,公式1以p代表类,p∈{phase_temp[1],phase_temp[2]},按照如下公式进行计算:
循环进行k轮后,以第一轮的phase_temp[1]为phase[1],以第一轮的phase_temp[2]为phase[2],将每轮的类phase_temp[1]、phase_temp[2]进行定相,p∈{phase1,phase2},
最终哪个相位的得分高,矩阵中该行reads则归为该相;
4)精细分相
对于定相后的矩阵,按照相位,分别拆分出两个新的矩阵;
新的矩阵采用步骤3)中所述方法,循环迭代进行,直到没有可用于分相的窗口;
跳出分相,得到多个一致性非常高的细分的矩阵;
5)矫正
细分的矩阵,进一步根据矩阵总深度矫正低深度比例的元素;
6)基因分型
合并矩阵,获得单体型序列;
所有的一致性序列均参与分型,得到每个型别的深度;当深度比例大于20或小于1/20,作为纯合子。
根据本发明的实施方式,所述步骤1)中的基因测序原始数据为三代基因测序原始数据。
进一步地,所述三代基因测序原始数据为pacbiosequel原始数据。
根据本发明的实施方式,所述步骤1)中的基因测序原始数据为hla基因测序原始数据。
根据本发明的实施方式,所述参考序列为数据库中相对应的最长一条序列。
根据本发明的实施方式,所述步骤1)中的长序列比对所用软件为blasr或graphmap软件。
根据本发明的实施方式,所述步骤1)中比对矩阵m的组成元素为:a,t,c,g,*,.;
根据本发明的实施方式,所述步骤1)中形成矩阵m1的方法为:矫正纯合位点深度小于5%的碱基;重新调整矩阵,去除m中均为*的列。
本发明的另一方面提供上述方法在在基因分型中的应用。
根据本发明的实施方式,所述基因分型为在三代测序基因分型。
根据本发明的实施方式,所述基因分型为hla基因分型。
有益效果
1、本方法可适合多态性高、重复序列复杂的序列;滑窗矫正分相,使序列真正的多态性彰显,去除了比对多样性和错误率的影响,从单体型的整体上权重相位。
2、随机选定多条序列分别作为参考,将随机错误率引起的杂合标签排除,达到精准的分相。
3、循环迭代分相排除尽可能多的原因,达到一致性最高的分相结果。
附图说明
图1为本发明基因分型算法流程图。
具体实施方式
下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例
本实施例针对人类白细胞抗原(humanleukocyteantigen,hla)基因进行三代单体型构建。
1、生成矩阵
pacbiosequel原始下机数据通过lima软件根据barcode拆分不同样品,并通过circularconsensussequencing算法进行序列之间的矫正获得circularconsensussequence(ccs)。将ccs与参考序列(针对每种hla基因,从ipd-imgt/hla数据库中取一条最长序列作为参考序列)进行长序列blasr或graphmap软件比对,采用samtools软件的tview命令,输出文本格式的比对矩阵,用m表示。分型流程算法见图1。
比对矩阵的组成元素为6个:a,t,c,g,*,.。用t表示,t∈{a,t,c,g,*,.}。
矫正“纯合位点(只有一种m的深度大于5%)”的深度小于5%的碱基;重新调整矩阵,去除m中均为“*”的列,形成粗矫正的矩阵m1。
2、分相
2.1按窗口编码归一化矩阵
编码m1,设置窗口长度与步长,每个窗口对应的矩阵横坐标、纵坐标分别为(i,j),用w[i]表示该窗口,w[i,j]表示窗口中的每条序列对应的矩阵元素,窗口长度为win,步长为step。
窗口每滑动一步,调整当前窗口的矩阵元素,调整规则以w[i]内序列相同的行,转换为同一种比对矩阵,w[i]则形成一个新的矩阵m[i]。
m[i]当前步长内的元素被确定,m[i]步长外的元素将继续进入下一个窗口继续调整。
整个扩增子经过一步一个窗口滑动,最终形成按照“步长和窗口”编码新的矩阵m2。2.2筛选杂合分相窗口
对于按照“步长和窗口”编码的矩阵m2,其每一步的窗口w[i],若窗口内每条矩阵元素w[i,j]对应的同种序列类型深度之和,只有一种超过了总深度的5%,则取消该窗口参与分相的资格。
否则若有大于5%的两种或以上序列类型,则标记为用来分相的窗口w[i]∈h;下一个被标记的窗口,和该窗口无交集。若同时相邻的窗口(上一个窗口的终点和下一个窗口的起点相邻)被标记为h,则所有的这些窗口合并为一个新的窗口w_combine[i]将重新作为一个窗口进行调整,即w_combine[i]内序列相同的行,转换为同一种比对矩阵,形成一个新的矩阵m_combine[i]。
被标记的杂合窗口形成了新的杂合矩阵h。
3、相位的确定
假设杂合矩阵h由n个被编码的窗口,矩阵的深度为d。
接下来将进行k轮循环分相。
随机选择k个小于d的数字,假设第t次随机选择了r,t∈random{1..k},r∈random{0..d},每个窗口的纵坐标为r的行,作为参考。每个窗口内均有两种最大深度的矩阵类型w[r,max,]、w[r,second,],这两种类型哪一种和参考的相似度高,将暂时归为一类phase_temp[1],另一种归为另一类phase_temp[2]。窗口内每行根据与w[r,max,]、w[r,second,]的相似度,划分类。
窗口内某行与phase_temp[1]、phase_temp[2]相似度等于“相同碱基/窗口长度”,完全一致时p=1。对于每一行计算最终属于phase_temp[1]、还是phase_temp[2]的得分,公式1以p
代表类,p∈{phase_temp[1],phase_temp[2]},按照如下进行计算:
循环进行k轮后,以第一轮的phase_temp[1]为phase[1],以第一轮的phase_temp[2]为phase[2],将每轮的类phase_temp[1]、phase_temp[2]进行定相p∈{phase1,phase2},
最终哪个相位的得分高,矩阵中该行reads则归为该相。
4、精细分相
对于定相后的矩阵,按照相位,分别拆分出两个新的矩阵。
新的矩阵采用以上方法,循环迭代进行,直到没有可用于分相的窗口。
跳出分相,得到多个一致性非常高的细分的矩阵。
5、矫正
细分的矩阵,将进一步根据矩阵总深度矫正低深度比例的元素。
6、基因分型
最终合并矩阵,获得单体型序列。
所有的一致性序列均参与分型,得到每个型别的深度;当深度比例大于20或小于1/20,作为纯合子。
按照以上方法得到分型结果与一代分型结果进行核查,准确率98.43%。
分型结果见表1。
表1
- 还没有人留言评论。精彩留言会获得点赞!