一种利用宏基因组和机器学习进行蜂蜜蜜源地追踪的方法
1.本发明涉及宏基因组测序、物种多样性分析、机器学习和生物信息学领域,具体涉及一种利用宏基因组和机器学习进行蜂蜜蜜源地追踪的方法。
背景技术:
2.在国内的蜂蜜市场中,西方蜜蜂采集产生的花蜜大多是单一花种蜂蜜(单花蜜),例如槐花蜜、油菜蜜等,而中华蜜蜂采集产生的往往是多花种蜂蜜(百花蜜)。西方蜜蜂相较于中华蜜蜂而言具有更好的产蜜能力,因此,在1900年前后被首次引入中国。并且随着西方蜜蜂的引入,一系列现代产业化的管理模式也随之引入,比如蜂群的日常清理和迁移。而大量地方上的蜂农对饲养中华蜜蜂依然具有更高的热情,尤其是在比较偏远的山区。尽管如此,西方蜜蜂的引入不可避免的致使中华蜜蜂种群受到威胁,导致在过去的100多年时间里种群密度的大幅下降。中华蜜蜂种群受到的影响多集中在农业活动密集的区域,而在人口稀疏的山区,西方蜜蜂对当地中华蜜蜂种群的影响有限。两种蜜蜂的生态竞争在蜂蜜生产和市场销售中却带来了意想不到的结果:消费者普遍认为来自农村和山区的蜂蜜产品,是通过原初的生产方式获得,是真正的绿色有机食品,因此相比于集中化生产的蜂蜜更加健康,这产生的最直接、最显著的结果就是,地方蜜蜂(也就是中华蜜蜂)的蜂蜜产品,尤其是在自然生态保护区收集的蜂蜜产品在市场中被标定为高端蜂蜜,定价也比西方蜜蜂的蜂蜜贵2
‑
8倍。而这种巨大的利润差异也刺激了一种新的蜂蜜产品欺诈行为:西方蜜蜂生产的蜂蜜或非自然生态地区生产的中华蜜蜂生产的蜂蜜贴上“高端蜂蜜”的标签在市场中鱼目混珠。
3.蜂蜜中的花粉特征为我们提供了一种追溯蜜源地的思路和方法:因为花粉粒往往通过工蜂在存储或取食花蜜/蜂蜜的过程中,又或者是养蜂人在采蜜过程中(例如通过挤压、过滤或离心)混入蜂蜜,而蜂蜜中花粉的组成从本质上能够反映当地开花植物物种多样性的组成。所以,来自同一地区,或者地理距离较近地区的蜂蜜理论上应该具有相似的花粉组成,而与地理距离较远的蜂蜜相比,其花粉的种类组成应该具有较大的区别。因此,利用不同地理区域开花植物组成的差异进行蜂蜜样品之间的比较,理论上就可以确定其蜜源地的差异。
4.然而,花粉鉴定并非易事。传统的鉴定方法,也被称为孢粉学,通过观察花粉粒的微观结构进行物种分类和鉴定。这种方法与其他基于形态特征进行物种鉴定的方式一样,需要消耗大量的人力,并且需要专业的知识储备。而且,因为花粉形态学数据的分辨率有限,也难以有效地区分很多植物类群内部的近缘种。除了上述的基于孢粉学进行蜂蜜的植物组成分析,有一些研究利用特定植物中独特的蛋白质或碳水化合物生物标记,基于生物化学的方法对特定物种进行分辨和鉴定,但是这种方法仅能检测特定的指示植物物种,无法一次性进行多种开花植物的分析和调查。
5.利用高通量测序的宏条形码技术进行花粉/蜂蜜中植物组成进行分析和鉴定。这些研究利用花粉中的核酸物质(dna)的序列特征进行物种鉴定和分析,进而得到蜂蜜中的
开花植物组成。虽然基于高通量测序手段的宏条形码分析手段能够识别蜂蜜中的粉源植物,但是不同的研究之间在对开花植物的物种鉴定的准确度水平上依然具有较大的差异,物种鉴定的差异多是由于不同研究所采用的遗传标记本身的分辨率、引物的通用性差异以及样品采集地区本底植物分子参考数据库不完整等问题造成。例如,在植物物种鉴定中常用的trnl
‑
uaa标准片段或rbcla通常只能够鉴定至科或属的水平。此外,宏条形码方法依赖于的目标dna片段的pcr扩增过程,包括matk,trnl
‑
uaa,rbcla,trnh
‑
psba等,而在pcr的扩增过程中,尤其是样品中同时存在多种远缘物种时,不同物种的扩增效率容易受到扩增偏向性的影响,即使是“通用引物”也会由于引物和模板之间的碱基差异而导致显著的扩增偏向性,这往往会影响物种分类单元的定量,甚至会影响特定分类单元的定性分析。而且宏条形码方法都很大程度上依赖于本地植物参考序列(即植物条形码或叶绿体基因组)的完整性和全面性。事实上,本地植物参考数据库的缺乏对所有基于高通量测序的花粉识别方法都提出了挑战。这一挑战对蜜源地追溯显得更为严峻,因为要得到每个目标蜜源地区域的关键指示物种,需要对当地植物物种进行全面彻底的调查,而鉴定指示物种的难度被多样地间的相互比较进一步放大。不可否认的是,目前对于世界上大多数地区,本地植物多样性的分子数据库匮乏的问题在短期内难以得到解决,而当我们的研究目标——蜂蜜产品来自于偏远不发达山区时,这个问题显得尤为突出。
技术实现要素:
6.本发明的目的是针对上述植物多样性分子数据库匮乏的问题,提供一种通过机器学习的方法对植物序列和蜜源地之间建立权重系数,以实现对蜂蜜样品蜜源地追踪的目的。该方法充分考虑蜂蜜蜜源地,尤其是偏远山区,植物物种的分子数据库匮乏的问题,分析蜂蜜中核酸物质的序列信息,进而得到蜂蜜中所包含的植物序列,然后通过机器学习的方法计算得到蜂蜜中植物源的核酸序列(不需要具体的物种鉴定结果)与其蜜源地的权重关系,并以此作为参考数据库进行蜂蜜样品的蜜源地追踪。该方法不依赖已有的蜜源地植物分子数据库,可以直接通过蜂蜜中植物序列分析和机器学习得到蜂蜜的蜜源地信息,对蜂蜜产品的鉴定和市场的规范有着极其重要的价值。
7.本发明的技术方案如下:
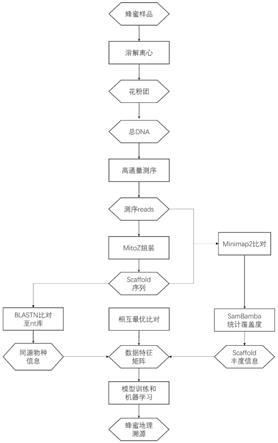
8.一种蜂蜜的蜜源地追踪方法,包括以下步骤:
9.1)收集已知蜜源地的蜂蜜样品,要求每个蜜源地包括多个样品以充分代表蜜源地的多样性和异质性。对蜂蜜样品进行花粉富集并进行核酸物质(dna)的抽提和纯化。
10.2)dna测序和质量过滤:对每个蜂蜜样品进行核酸序列的测定,并进行测序数据常规质量过滤,包括低质量序列的去除和测序接头序列的去除。
11.3)对质量过滤之后的测序数据进行组装,并通过将所有的原始测序数据比对回组装结果以得到组装序列的丰富度信息。为保证后续序列分析的可靠性,本方法只保留组装长度大于500bp以及丰富度大于3的组装序列。
12.4)对步骤3)中组装得到的序列进行物种信息注释,将组装序列比对到ncbi的核苷酸序列数据库(nt库),并记录最优比对结果的物种信息。为保证物种信息的可靠性,比对覆盖度达到序列本身长度80%以上,或者大于1,000bp的比对结果才认为是有效的比对结果。根据物种的注释信息,提取植物源的序列进行后续的分析。
13.5)对步骤4)中得到的植物源的序列进行样品间的两两比对,根据两两比对的结果首先将同一蜂蜜样品中来自同一物种的组装序列整合,并记录不同样品间共享的序列信息。共享的序列需要满足序列相似度大于等于98%,比对总长大于等于1,000bp。
14.6)对步骤5)中得到的样品间共享的序列信息分析整合,构建不同蜂蜜样品的数据矩阵,其中矩阵的变量为所有蜜蜂样品的植物源代表序列,蜂蜜样品含有这条序列,或者共享该序列,则该蜂蜜样品这条植物源序列变量的值为1,不符合上述情况则计值为0。同时,对蜂蜜样品进行地理位置变量的定值,蜂蜜样品根据其蜜源地得到不同地理位置的概率值,每个蜂蜜样品在其蜜源地的概率值为100%,其他地理位置的概率为0%。
15.7)对步骤6)中得到的数据矩阵进行基于神经网络模型的数据训练,其中植物源序列变量作为神经网络的输入层(input layer),而地理位置变量作为神经网络的输出层(output layer),同时根据输入层变量的数目决定隐含层(hidden layer)的数量和神经元的数目。
16.8)对待检测的蜂蜜样品提供输入层变量值,用步骤7)中得到的训练模型,进行蜜源地概率的估算。其中概率值最高的地理位置为其的蜜源地。
17.上述步骤1)中应尽量保证采集的蜂蜜样品来自不同的蜂巢和地点,并且在蜂蜜样品的选取中应该保证蜜源地的准确性和可追溯性,这样在进行后续蜂蜜样品的植物源核酸序列确定过程中可以保证尽量完整和准确地覆盖当地的植物源。
18.优选的,步骤1)中使用当地蜂农采集的原蜜,步骤2)中每个蜂蜜样本测序量平均为4
‑
5g原始数据。
19.优选的,上述步骤3)中,对于序列的组装应该采用宏基因组优化的组装算法,可以使用mitoz软件包中的组装算法进行序列的组装,并设置k
‑
mer的长度大于50,在edge丰度方面设置最小值为3。然后利用minimap2将原始测序序列比对到组装序列上,通过sambamba统计每条组装序列的覆盖度信息,并对样本数据进行均一化(以一百万测序序列为基数)处理,进而得到组装序列的丰富度信息。
20.上述步骤4)中,对于每个蜂蜜样品中的植物源序列,选取丰富度最高的50即可,亦或可以选取丰富度占比总计99%以上的序列即可以提高数据矩阵变量的有效性,进而提高神经网络数据训练的效率和准确性。同时,上述步骤6)中构建数据矩阵时,可以进一步过滤植物源序列变量,过滤掉蜜源地的无效变量,即在同一蜜源地中只单次出现的变量。
21.上述步骤8)中,对于每个待测样品,可以独立运行多次检测,并要求概率的最高值10倍高于次高概率值时,认为其是一次有效检测,保证检测的可靠性。
22.本发明结合蜂蜜的宏基因组测序和机器学习的方法,进行蜂蜜蜜源地的追踪和归属分析。该方法相对于现有的基于测序的蜂蜜蜜源地追踪的方法而言,可以直接利用蜂蜜中获取的植物源组装序列作为特征变量,不依赖于蜜源地本底的植物分子数据库,而且机器学习的方法避免了大量的人工成本去寻找和定位地理区域代表性的特征植物源序列,能够通过已知数据集的训练得到各植物源序列对应蜜源地的权重和贡献比例,因此本发明,相较于现有的方法,在前端数据库准备方面和后续特征值提取方面都具有更高的可行性。本发明为蜂蜜的鉴定和蜂蜜产业的规范着重要的意义。
附图说明
23.图1.本发明蜂蜜蜜源地追踪方法的流程图。
24.图2.具体实施方式中蜂蜜样品的地理位置信息,其中,左下角放大图中详细展示了四川的采样点分布。
25.图3.交叉验证实验中同一蜜源地蜂蜜样品的归属地概率分布图,其中,柱状图展示了特定地理来源(x轴)的蜂蜜样品在交叉验证测试中的结果分布,不同地理位置的概率分布通过不同颜色深度区分,交叉验证结果符合其来源地的结果显示为深色,而非蜜源地则显示为浅色;其中,qinghai:青海;beijing:北京;jilin:吉林;aba:阿坝;laohegou:老河沟;guanba:关坝。
26.图4.交叉验证实验中独立蜂蜜样品的归属地概率分布图,其中,每个蜂蜜样品的单次交叉检验将产生7个点位,对应于7个不同蜜源地的概率分值,蜜源地通过不同的颜色深度进行区分,交叉验证结果符合其来源地的结果显示为深色,而非蜜源地则显示为浅色;其中,qinghai:青海;beijing:北京;jilin:吉林;aba:阿坝;laohegou:老河沟;guanba:关坝.
具体实施方式
27.以下所述是本发明的更加细致的实施描述,其中的参数以及具体实施细节用以解释本发明的可行性及实施效果,并不构成对本发明的限定。
28.本实施例包括28份蜂蜜样品,除了1个产自西方蜜蜂的蜂蜜样品外,其余27个蜂蜜样品全部产自于中华蜜蜂。其中有9个样品来自于中国四川省老河沟自然保护区(lhg),该保护区属于大熊猫的自然栖息地范围。为了检验利用当地的植物组成是否能够准确的区分其他地区采集的蜂蜜样品和来自于老河沟地区的蜂蜜样品,采集了与老河沟临近的两个样点(关坝
‑
gb和王朗
‑
wl),以及中国其他不同地区5个样点的蜂蜜样品,包括阿坝
‑
ab、北京
‑
bj、吉林
‑
jl、陕西
‑
sx和青海
‑
qh。老河沟、关坝、王朗、阿坝、北京、吉林、陕西和青海样点的蜂蜜样品数分别为9、2、1、3、4、3、3和3(图2)。
29.1.样本核酸物种获取及测序
30.将每个样品分别取50g的蜂蜜,分别称量至4个50ml无菌离心管中(4
×
12.5g),并向每个管中添加30ml超纯水。然后将蜂蜜在65℃的水浴中溶解15分钟。之后,将混合物以12,000rpm离心15分钟,弃上清。最后,将花粉沉淀从4个不同的离心管取出合并至1个离心管中,并重复上述溶解和离心步骤1次。最终的沉淀物用于dna提取。使用改良的wizard方法(soares,amaral,oliveira,&mafra,2015)对每个蜂蜜样品约0.5g沉淀物(混合花粉粒)进行dna提取。每个蜂蜜样品的200ng dna用于构建插入片段大小为350bp的测序文库(dual
‑
indexed,双标记),随后在illumina hiseq x ten平台上进行150pe测序。
31.2.序列组装和注释
32.我们使用mitoz的组装模块对每个样本进行组装以获得scaffold序列(k
‑
mer=31,minimum_edge_depth=3,minimum_output_length=500)。接着,利用minimap2将原始测序序列比对到组装序列上,然后通过sambamba统计每条组装结果序列的覆盖度信息,并对样本数据进行均一化(以一百万测序序列为基数)处理,进而得到组装结果的丰富度信息。随后,我们利用blastn将组装结果与nt库(ncbi)进行比对,以获得组装序列的同源物种
信息。同时,我们在组装序列与nt库的比对结果中提取得到公共数据库中与之最近缘序列的相似性得分和物种分类信息。
33.3.序列矩阵特征构建
34.我们使用了一种相互最优比对的方法(reciprocal blast)来对蜂蜜样品间的序列组成进行两两比较,本方法通过对每个蜂蜜样品的组装序列与其他所有蜂蜜样品的组装序列进行blast比对,当同一样本的多条组装序列可以映射到其他样品中同一序列的不同区域,且相似度≥98%,覆盖率≥80%时,我们认为这些序列来自于同一物种,并将这些序列以及其丰度合并以进行下游分析。随后,为了进一步提高计算效率,对单个蜂蜜样本我们选取其丰度最高的50条植物代表序列进行后续的分析。在蜂蜜样品间的比较分析中,不同样品序列间blast比对结果相似度≥98%且比对长度≥1,000bp被定义为同一植物物种,在数据矩阵中显示为1,否则则记录为0。最后,单一蜂蜜样品中的特异性序列,即在地理区域仅出现一次的组装序列,我们认为其不含有任何地理性的有效信息,将会在随后的分析中删除。
35.4.机器学习模型训练
36.我们利用“neuralnet”r包中的弹性反向传播(resilient backpropagation)的人工神经网络算法对数据特征矩阵进行训练,其中在数据训练过程中采用了单隐含层(hidden layer)和半数于序列特征变量的神经元(neuron)数目。神经网络的计算方法将多个变量(如本研究中上述的植物来源的序列)的作为神经网络的输入层(input layer),然后对所有的蜂蜜样品,我们根据其地理归属,将其本身地理变量的概率设定为100%,将其他地理变量的概率设定为0%,并把这些概率值作为神经网络的输出层(output layer)。神经网络算法通过隐含层中的若干神经元(在本案例中为植物源序列变量数目的1/2)以估算训练数据集中每个序列变量的特征值和权重,最终通过最优拟合得到输出层中每个蜂蜜样品的地理归属概率,而最优拟合是通过隐藏层中所包含的数学函数实现的,这些数学函数对输入数据矩阵中每一个观察值执行非线性转换(本方法中是resilient backpropagation算法)以得到该观察值的期望结果。
37.5.留一法交叉验证
38.通过留一法交叉验证(leave
‑
one
‑
out cross
‑
validation,loocv)以评估训练模型的准确性和机器学习的表现。留一法交叉验证,是穷尽交叉验证的方法之一,顾名思义就是抽取一个观察结果作为验证集,而利用其余的观察结果作为训练集,对数据集合中所有观察结果逐一进行验证,以达到评估神经网络算法准确性的目的。在每一次交叉验证测试中,通过训练数据集的机器学习,得到数据模型中关于输入层变量的权重和特征信息以及隐含层中数学函数的参数,进而对验证数据中变量进行计算,得到其在多个地理位置的分配概率,概率最大的地理位置被认为是该测试样品的蜜源地,同时我们要求测试样品地理归属的最大概率值超过第二概率值的10倍,才将其作为一次有效的交叉验证。例如,我们从全部数据集中抽掉一个老河沟的样本,进而得到一个训练集,其中包括其余8个老河沟样品以及其他所有的蜂蜜样品。然后,我们可以利用此训练集进行基于神经网络的机器学习,并通过训练好的模型对上述抽掉的一个老河沟样本计算其多个地理位置的分配概率。我们一共进行了1,000次的交叉验证测试,并将结果记录以用于后续的统计。因为王朗仅包含一个样品,因此将不作为测试样品进行交叉验证。
39.6.溯源蜂蜜样品的蜜源地
40.综上过机器学习得到的训练模型,以及以此训练模型进行分析得到的多个蜜源地归属概率的结果即可进行每个蜂蜜样品的蜜源地追踪。图3为来自同一蜜源地的所有蜂蜜样品归属地的归属地追踪概率结果,其中其不同地理位置的概率以柱状图进行总结和展示,以不同的颜色区分不同的地理位置。例如,来自阿坝的蜂蜜样品在交叉检验中得到其归属于阿坝的概率分值较高(89.38
‑
99.74%),在归属于其他6个地理位置的概率得分较低。值得注意的是,样品h0003、h0018和h0028分别贡献了老河沟、陕西和北京的大部分异常值。图4为每个蜂蜜样品独立进行其蜜源地归属概率的总结和展示,每一个点代表一次独立的交叉验证结果,以不同的颜色的区分不同的地理位置。例如,h0028样品的一次交叉检验将会在x轴为h0028的位置产生7个不同蜜源地的概率分值点。可以看出,使用本发明方法能够对几乎所有的蜂蜜样品准确的进行蜜源地追踪,蜜源地分辨率能够达到百公里以内,比如关坝和老河沟样点之间只有约40km的距离。
41.以上所述仅作为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之类的所做的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1