一种非贵金属氮掺杂石墨烯电催化剂的制备及应用的制作方法
本发明涉及一种非贵金属氮掺杂石墨烯电催化剂的制备方法及其应用。
背景技术:
随着能源环境问题日益凸显,氢氧燃料电池由于其能量转换效率高、环境友好等优点受到了专家学者的关注,而电池阴极反应氧气还原反应(orr)最理想的催化剂是铂碳催化剂,但是铂昂贵的价格和有限的储量极大的限制了燃料电池的商业化。由于较高的活性和较低的成本,以氮掺杂石墨烯为基础的非贵金属催化剂成为了研究热点(z.-h.sheng,l.shao,j.-j.chen,w.-j.bao,f.-b.wangandx.-h.xia,acsnano,2011,5,4350-4358)。目前,氮掺杂石墨烯氧还原电催化剂的制备方法主要是将氮源与氧化石墨烯物理混合,所用氮源一般为尿素、双氰胺或三聚氰胺等,然后加入还原剂将氧化石墨烯还原,然而此方法容易使石墨烯堆叠,且需要用到pdda等表面活性剂,使实验步骤繁琐(q.q.wang,l.wang,g.p.liandb.x.ye,talanta,2017,164,323-329.)。
由于金属-有机骨架(metalorganicframework,mof)结构中含有碳源(有机配体)和金属源(金属或金属簇)物质,以mofs为前驱体合成先进功能材料,如纳米氮掺杂碳材料和金属氧化物纳米材料,是目前mofs化学以及新功能材料研究领域一个新的热点。yao等人(x.zhaoet.al.,j.mater.chem.a,2014,2,11666–11671)以nh2-mil-53(al)为前驱体一步法合成非金属氮掺杂微孔碳材料,得到的催化剂在载量0.061mg/cm-2时碱性体系下半波电位与pt/c相差约60mv,然而该催化剂制备过程中需用到剧毒腐蚀性hf酸除去al,操作过程危险且不符合原子经济型制备原则。song等人(h.huanget.al.,j.mater.chem.a,2015,3,4976–4982)以fe-mil-88b(醋酸作溶剂)、双氰胺和蔗糖为原料,热解后得到fe3c/氮掺杂石墨烯催化剂,碱性条件下起始电位高于商业化pt/c10mv,然而其制备过程需加入表面活性剂且在电极表面的担载量较高。
世界专利wo2017042564a1公开了一种方法,以醋酸铁、1,10-菲罗林以及自制的cat-28、cat-19、mof-5三种含zn且内孔体积大于0.7cm3/g的mof为前驱体,球磨并在nh3下热解后得到非贵金属氧还原催化剂。该制备方法的制备步骤较简单,然而需要加入菲罗林和nh3两类氮源,且全电池电化学活性不高。世界专利2017117410a1公开了一种方法,通过第一过渡金属化合物和第一有机配体之间的机械化学反应形成第一mof产物后,加入第二过渡金属和第二有机配体的第二mof产物,分别在ar和nh3下两步热解后得到的催化剂在0.6mg/cm2的担量下,酸性体系半电池的起始电位可达0.85v。但该方法需要用到球磨,球磨过程可能引入杂质导致金属物种不纯净,而且需要两步热解,增加合成步骤。中国专利201610873701公开了一种方法,以尿素为氮源掺杂石墨烯,在水热条件下使n掺杂的go表面负载钴的化合物,再在水热条件下用水合肼还原得到氮掺杂石墨烯负载钴复合材料,在碱性溶液中氧还原催化性能一般,然而需要用到水合肼以及减压抽滤等步骤。中国专利201610873701.5公开了一种方法,使用双金属氧化物作为前体,利用类水滑石化合物的记忆效应,氧化石墨烯和碳氢化钙类水滑石化合物,然后在还原条件下将化合物掺杂氮化碳纳米片,得到掺杂氮的石墨烯/铁氰化钴类化合物,该催化剂在碱性条件下同时具有对析氧反应(oer)和氧还原反应(orr)的催化活性,然而性能最佳的实施例催化剂半波电位与商业化pt/c仍相差60mv。
技术实现要素:
本发明所要解决的技术问题是提供一种简单、低成本地制备非贵金属氮掺杂石墨烯电催化剂的方法。
本发明包括以下步骤:
将石墨烯分散到有机溶剂中,超声分散数小时后分别加入非贵金属-mof的金属硝酸或醋酸盐和有机配体,反应、离心、冷冻干燥后,热解可得到担载型的非贵金属氮掺杂石墨烯电催化剂。
有机溶剂为甲醇、乙醇、异丙醇、乙二醇或聚乙二醇等中的一种或二种以上。
非贵金属-mof前驱体为mof-5(zn)、zif-8(zn)、zif-67(co)、mil-88b(fe)或mil-101(fe)中的一种或二种以上;其中zif为沸石咪唑骨架(zincimidazolateframework)的简称,mil为拉瓦锡研究所材料(materiauxdel’institutelavosier)的简称。
石墨烯在有机溶剂中的浓度为0.1-10mg/ml,优选0.1-5mg/ml。
石墨烯、非贵金属-mof的金属硝酸或醋酸盐、有机配体在有机溶剂中混合后的质量比为20-130mg:0.1-5g:0.1-5g。
反应温度为10-200℃,优选10-150℃,反应时间为5-48小时,优选5-30小时。
热解的温度为300-1200℃,优选500-1000℃;热解的时间为5分钟-5小时,优选1-3小时。
所述非贵金属氮掺杂石墨烯电催化剂可作为阴极催化剂在碱性燃料电池中应用。
本发明采用低成本的非贵金属盐作为金属源,石墨烯为碳载体,高比表面、多孔的mof材料为前驱体,提高了催化活性;制备过程无需额外添加氮源、无需使用表面活性剂、还原剂水合肼等,即可得到氧还原电催化剂;制备氮掺杂石墨烯过程并未采用牺牲元素,属于原子经济型制备方法;mof前驱体可以选择的种类及组成较为广泛,使本发明有较大的适用范围。
附图说明
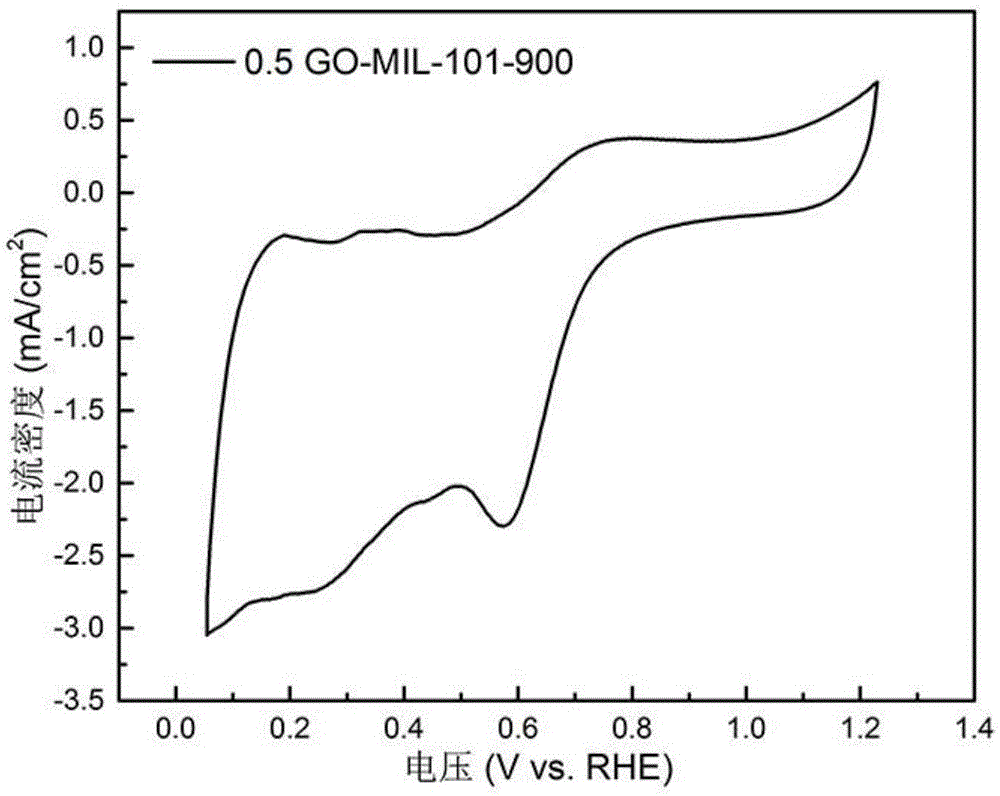
图1与图2分别为实施例一制备的0.5go-mil-101-900催化剂通过旋转圆盘电极(rde)测试得到的循环伏安曲线与氧还原极化曲线。
图3为实施例三制备的2go-mof-5-900催化剂的sem图;
图4为实施例三制备的2go-mof-5-900催化剂的xrd图。
图5与图6分别为实施例五制备的1go-zif-67-900催化剂在旋转圆盘电极(rde)测试中加速衰减测试与电流-时间曲线图。测试用电解质均为o2饱和的0.1mol/lkoh水溶液,均在室温下进行,电极上金属担量为400μg/cm2。加速衰减测试扫速为100mv/s,扫描范围0.6-1.0vvs.rhe。电流-时间曲线测试恒定电位0.8v,rde转速为1600rpm,在第180秒加入甲醇使得电解质甲醇体积分数为5%。
图7与图8分别为实施例七制备的1.6go-mof-5-900催化剂的sem图;以及通过旋转圆盘电极(rde)测试得到的循环伏安曲线与氧还原极化曲线。
图9为实施例八制备得到的0.5go-zif-67-900催化剂的sem图;
图10为实施例八制备得到的0.5go-zif-67-900催化剂tem图;
图11为实施例八制备得到的0.5go-zif-67-900催化剂的nldft孔径分布图。
图12为实施例八制备的0.5go-zif-67-900催化剂在旋转圆盘电极(rde)测试中线性扫描伏安图。线性扫描测试用电解质为o2饱和的0.1mol/lkoh水溶液,扫速为10mv/s,正向扫描,rde转速为1600rpm,扫描范围0.2-1.2vvs.rhe。测试均在室,温下进行,电极上催化剂担量为400μg/cm2。
图13为比较例一所采用的zif-67-900催化剂相应的前驱体zif-67的sem图;
图14为前驱体mof-5的sem图(w.bao,et.al.,electrochimicaacta,2014,127,342–348);
图15为前驱体zif-67的物理吸附曲线(y.bai,et.al.,chinesejournalofcatalysis,2016,37,1127–1133)。
图16为比较例一制备的zif-67-900催化剂在旋转圆盘电极(rde)测试中线性扫描伏安图。线性扫描测试用电解质为o2饱和的0.1mol/lkoh水溶液,扫速为10mv/s,正向扫描,rde转速为1600rpm,扫描范围0.2-1.2vvs.rhe。测试均在室温下进行,电极上催化剂担量为400μg/cm2。
具体实施方式
实施例一:
1.取62.5mg石墨烯于单口烧瓶中,加入125ml乙醇,超声分散5个小时后,使得go的浓度为0.5mg/ml,加入0.6488gfecl3·6h2o及0.3967g对苯二甲酸;150℃下搅拌反应24小时后,离心冷冻蒸干后得到浅灰色粉末。
2.将上述浅灰色粉末置于管式炉石英舟内热解,通入流速为80ml/min的氩气,升温速率为每分钟5℃,在900℃保持2小时后自然降温,得到0.5go-mil-101-900催化剂。
图1与图2分别为实施例一制备的0.5go-mil-101-900催化剂通过旋转圆盘电极(rde)测试得到的循环伏安曲线与氧还原极化曲线。
实施例二:
1.取250mg石墨烯于单口烧瓶中,加入125ml乙醇,超声分散5个小时后,使得go的浓度为2mg/ml,加入1.1900gzn(no3)2·6h2o及1.6405g2-甲基咪唑;室温下搅拌反应24小时后,离心冷冻蒸干后得到灰白色粉末。
2.将上述灰白色粉末置于管式炉石英舟内热解,通入流速为80ml/min的氩气,升温速率为每分钟5℃,在600℃保持2小时后自然降温,得到2go-zif-8-600催化剂。
实施例三:
1.取160mg石墨烯于单口烧瓶中,加入80mldmf,超声分散5个小时后,使得go的浓度为2mg/ml,加入7.0gzn(no3)2·6h2o及1.3935g对苯二甲酸;120℃下搅拌反应24小时后,离心冷冻蒸干后得到灰黑色粉末。
2.将上述灰黑色粉末置于管式炉石英舟内热解,通入流速为80ml/min的氩气,升温速率为每分钟5℃,在900℃保持2小时后自然降温,得到2go-mof-5-900催化剂。
图3为实施例三制备的2go-mof-5-900催化剂的sem图;图4为实施例三的2go-mof-5-900催化剂的xrd图。
图14为前驱体mof-5的sem图,sem图显示该颗粒为尺寸为5μm的正方体,与文献w.bao,et.al.,electrochimicaacta,2014,127,342–348中的mof-5形貌相一致,说明成功合成了前驱体mof-5;
实施例四:
1.取62.5mg石墨烯于单口烧瓶中,加入125ml乙醇,超声分散5个小时后,使得go的浓度为0.5mg/ml,加入1.1900gzn(no3)2·6h2o及1.6405g2-甲基咪唑;室温下搅拌反应24小时后,离心冷冻蒸干后得到浅紫色粉末。
2.将上述浅黄色粉末置于管式炉石英舟内热解,通入流速为80ml/min的氩气,升温速率为每分钟5℃,在900℃保持2小时后自然降温,得到0.5go-zif-8-900催化剂。
实施例五:
1.取125mg石墨烯于单口烧瓶中,加入125ml乙醇,超声分散5个小时后,使得go的浓度为1mg/ml,加入1.1640gco(no3)2·6h2o及1.6405g2-甲基咪唑;室温下搅拌反应24小时后,离心冷冻蒸干后得到浅紫色粉末。
2.将上述浅紫色粉末置于管式炉石英舟内热解,通入流速为80ml/min的氩气,升温速率为每分钟5℃,在900℃保持2小时后自然降温,得到1go-zif-67-900催化剂。
图5与图6分别为实施例五制备的1go-zif-67-900催化剂在旋转圆盘电极(rde)测试中加速衰减测试与电流-时间曲线图。测试用电解质均为o2饱和的0.1mol/lkoh水溶液,均在室温下进行,电极上金属担量为400μg/cm2。加速衰减测试扫速为100mv/s,扫描范围0.6-1.0vvs.rhe。电流-时间曲线测试恒定电位0.8v,rde转速为1600rpm,在第180秒加入甲醇使得电解质甲醇体积分数为5%。
实施例六:
1.取62.5mg石墨烯于单口烧瓶中,加入125ml乙醇,超声分散5个小时后,使得go的浓度为0.5mg/ml,加入1.1640gco(no3)2·6h2o及1.6405g2-甲基咪唑;室温下搅拌反应24小时后,离心冷冻蒸干后得到浅紫色粉末。
2.将上述浅紫色粉末置于管式炉石英舟内热解,通入流速为80ml/min的氩气,升温速率为每分钟5℃,在700℃保持2小时后自然降温,得到0.5go-zif-67-700催化剂。
实施例七:
1.取168mg石墨烯于单口烧瓶中,加入106mldmf,超声分散5个小时后,使得go的浓度为1.6mg/ml,加入7.0001gzn(ch3coo)2·2h2o及1.3934g对苯二甲酸,在125℃下回流24小时后离心得到黑色浆料,离心洗涤,冷冻干燥后得到黑色粉末。
2.将上述黑色粉末置于管式炉石英舟内热解,通入流速为80ml/min的氩气,升温速率为每分钟5℃,在900℃保持2小时后自然降温,得到1.6go-mof-5-900催化剂。
图7与图8分别为实施例七制备的1.6go-mof-5-900催化剂通过旋转圆盘电极(rde)测试得到的循环伏安曲线与氧还原极化曲线。
实施例八:
1.取62.5mg石墨烯于单口烧瓶中,加入125ml乙醇,超声分散5个小时后,使得go的浓度为0.5mg/ml,加入1.1640gco(no3)2·6h2o及1.6405g2-甲基咪唑;室温下搅拌反应24小时后,离心冷冻蒸干后得到浅紫色粉末。
2.将上述浅紫色粉末置于管式炉石英舟内热解,通入流速为80ml/min的氩气,升温速率为每分钟5℃,在900℃保持2小时后自然降温,得到0.5go-zif-67-900催化剂。
图9为实施例八制备得到的0.5go-zif-67-900催化剂的sem图;图10为实施例八制备得到的0.5go-zif-67-900催化剂tem图;图11为实施例八制备得到的0.5go-zif-67-900催化剂的n2物理吸附曲线。
图12为实施例八制备的0.5go-zif-67-900催化剂在旋转圆盘电极(rde)测试中线性扫描伏安图。线性扫描测试用电解质为o2饱和的0.1mol/lkoh水溶液,扫速为10mv/s,正向扫描,rde转速为1600rpm,扫描范围0.2-1.2vvs.rhe。测试均在室温下进行,电极上催化剂担量为400μg/cm2。
比较例一:
将0.1gzif-67紫色粉末置于管式炉石英舟内热解,即无石墨烯担载;通入流速为80ml/min的氩气,升温速率为每分钟5℃,在900℃保持2小时后自然降温,得到zif-67-900催化剂。
图13为比较例一所采用的zif-67-900催化剂相应的前驱体zif-67的sem图;图15为前驱体zif-67的物理吸附曲线,根据吸附曲线得出相应的bet比表面积为1009.3m2/g,与文献y.bai,et.al.,chinesejournalofcatalysis,2016,37,1127–1133中的值相近,说明成功合成了前驱体zif-67。
图16为比较例一制备的zif-67-900催化剂在旋转圆盘电极(rde)测试中线性扫描伏安图。线性扫描测试用电解质为o2饱和的0.1mol/lkoh水溶液,扫速为10mv/s,正向扫描,rde转速为1600rpm,扫描范围0.2-1.2vvs.rhe。测试均在室温下进行,电极上催化剂担量为400μg/cm2。
对比其与实施例八制备的0.5go-zif-67-900催化剂的活性可以看出,没有go担载的zif-67-900催化剂(比较例一)的催化活性低于有go担载的催化剂,说明本发明go的担载对于催化剂活性的提高有一定的作用。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种纺织纤维/石墨烯/SrTiO<sub>3</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiVO<sub>4</sub>/Bi<sub>2</sub>O<sub>3</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiVO<sub>4</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiVO<sub>4</sub>/Bi<sub>2</sub>WO<sub>6</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiVO<sub>4</sub>/BiPO<sub>4</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/Bi<sub>2</sub>MoO<sub>6</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/Bi<sub>2</sub>WO<sub>6</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/Bi<sub>2</sub>MoO<sub>6</sub>/BiPO<sub>4</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/Bi<sub>2</sub>GeO<sub>5</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiOBr/BiOI复合环境催化材料的制备方法