纳米相分离的固体聚合物电解质薄膜及其制备方法和应用与流程
1.本发明涉及储能技术领域,特别涉及一种纳米相分离的固体聚合物电解质薄膜及其制备方法和应用。
背景技术:
2.锂离子电池自1991年由日本索尼公司产业化后,便在各类3c产品、动力电池、大规模储能等领域发挥了至关重要的作用。随着国民经济的发展,人们对于锂离子电池提出了更高的要求,包括高能量密度和高安全性等。因此,开发兼具高能量密度和高安全性能的锂离子电池来满足新时代的要求迫在眉睫。
3.目前的锂离子电池多采用酯类、醚类等液态有机电解液作为电解质,但液态有机电解液存在漏液、易挥发、容易燃烧甚至爆炸等安全问题。另外,电池在循环过程中,锂枝晶的生成会也刺穿隔膜,引发安全事故。因此人们将目光转移到了固态电解质上。通常来说,固态电解质的热稳定性、化学稳定性、电化学稳定性和机械强度一般均优于都液体电解质。而且固态电解质的使用从理论上说可以从根本上消除安全隐患。同时固态电解质的电化学稳定性窗口可以高达5v,因此可用于高压正极材料,进而提高电池的能量密度。此外,固态电解质也能够实现高的锂离子传输系数和更好的机械强度,从而促进更加均匀的锂金属沉积。
4.目前,常见的固态电解质包括无机固体电解质、凝胶聚合物电解质和固体聚合物电解质。在固体电解质中,固体聚合物电解质具有较高的锂离子电导率和电极/电解质接触界面,可应用于锂离子电池和锂金属电池等固态电池。但是,聚合物的结晶性导致机械性能和离子电导率之间的矛盾。传统的基于线性聚乙烯基醚(peo)的固体电解质在室温下结晶度高但是锂离子电导率低,通过添加无机填料或者有机增塑剂可以提高聚合物固体电解质的离子电导率,但是会导致结晶度降低而影响固体聚合物电解质的力学性能;化学交联可以有效提高聚合物的机械强度,但是通常会形成结晶度高、锂离子电导率低的刚性聚合物。有文献已经证明聚合物电解质分子中无定形区可以有效地传导锂离子,增加聚合物电解质分子柔性链段的占比则可以有效地增加体系内无定形区域的占比,从而促进离子的输运过程。根据“刚柔并济”的设计理念通过将刚性主链与柔性侧链相结合可以有效地缓解固态聚合物电解质高机械性能与高室温离子电导率难以兼具的矛盾。在前期的工作中,我们也设计和合成了一种硫醇枝化结构的聚合物固体电解质(m-s-pegda),由mofs(uio-66)、四(3-巯基丙酸)季戊四醇酯(petmp)和长链聚(乙二醇)双丙烯酸酯(pegda)经化学交联形成。由于三者之间的协同作用,m-s-pegda具有优异的力学性能、高的离子电导率和低的界面电阻(202010028271.3)。
5.聚乙二醇是一种常见的可加工的peo基聚合物,其具有成本低、生物相容性高等优点,将其用作聚合物电解质基体,具有较低的玻璃化转变温度以及较高的电化学稳定窗口,因此被广泛用于聚合物电解质的合成。但目前基于peo的固态聚合物电解质依然难以满足锂金属电池的实际使用需求。存在的主要问题是高室温离子电导率与优秀的机械性能难以
兼得,因为高离子电导率往往意味着较差的机械强度。与此同时,基于聚醚的聚合物电解质(例如peo和pdo等)在充电电压超过4v的条件下容易发生电化学氧化降解,这极大地限制了它们在高能量密度锂电池中的应用。上述问题极大的限制了固态电解质的实际应用和锂金属电池的发展。
技术实现要素:
6.本发明提供一种纳米相分离的固体聚合物电解质薄膜及其制备方法和应用,通过聚合物分子结构设计实现聚合物固体电解质机械性能和锂离子电导率的解耦;得到的纳米相分离固体聚合物电解质同时兼具优异的机械性能、高的离子电导率和宽的电化学窗口。
7.本发明的技术方案是,一种纳米相分离的固体聚合物电解质薄膜,其特征在于,以聚苯乙烯-马来酸酐无规共聚物为刚性主链,在主链上接枝小分子量的聚乙二醇链段,利用主链和侧链在机械性能和化学亲和性的差异,通过溶液浇铸法得到具有纳米相分离结构的聚合物固体电解质薄膜;其中,聚苯乙烯-马来酸酐无规共聚物与聚乙二醇的质量比为1:1~4。
8.进一步地,其中聚苯乙烯-马来酸酐无规共聚物的分子量为1500~20000,通过控制溶剂的添加量来控制无规共聚物的分子量;聚乙二醇的分子量为400~6000,马来酸酐的浓度为0.16-0.5mmol/ml之间,。
9.进一步地,溶液浇铸过程中,加入有电解质锂盐,锂盐为litfsi,lifsi,libob,lifob中的一种或几种。优选为litfsi和lidfob的摩尔比1:1的复配锂盐。
10.锂盐添加量为聚乙二醇接枝马来酸酐-苯乙烯无规共聚物质量的5~35%。
11.本发明还涉及纳米相分离的固体聚合物电解质薄膜的制备方法,具体包括以下步骤:
12.s1、苯乙烯-马来酸酐无规共聚物的制备将苯乙烯加入有氮气保护的容器中,然后将马来酸酐和引发剂分散于溶剂中,滴加入苯乙烯中,同时加热引发聚合反应,反应完毕后冷却、抽滤、干燥后得到苯乙烯-马来酸酐无规共聚物;
13.s2、将合成的苯乙烯-马来酸酐无规共聚物、小分子量聚乙二醇和催化剂溶解于溶剂中,在氮气保护下回流加热反应,再利用氢氧化钠的醇溶液中和,冷却、抽滤、干燥得到聚乙二醇接枝马来酸酐-苯乙烯无规共聚物;
14.s3、聚乙二醇接枝马来酸酐-苯乙烯无规共聚物用溶剂溶解,加入锂盐加热搅拌得到分散均匀的铸膜液,铸膜液静置之后进行超声脱泡处理,再将处理好的铸膜液流延成型,真空干燥后即得到聚合物固体电解质膜。
15.进一步地,s1中,苯乙烯和马来酸酐的物质的量比在5~15:1;优选为9:1。所述有机溶剂为苯、甲苯、丙酮、氯化烃、碳酸二甲酯、或乙酸异戊酯中的一种或几种;所述引发剂为过氧化二异丙苯dcp,bibp,bpo或aibn中的一种;反应温度为60~150℃;反应时间为1~5小时。
16.进一步地,s2中,苯乙烯-马来酸酐无规共聚物与小分子量聚乙二醇的质量比为1:1~4。优选为1:2。所述溶剂为四氢呋喃、二氯甲烷中的一种。所述聚乙二醇接枝苯乙烯-马来酸酐无规共聚物(r-sma-g-peg)是通过酯化反应将小分子量的聚乙二醇接枝到聚苯乙烯-马来酸酐无规共聚物的马来酸酐单元,清洗干燥后得到白色固体。
17.进一步地,s3中,所用溶剂为n,n-二甲基乙酰胺,乙腈,碳酸乙烯酯,碳酸丙烯酯中的一种或者几种。
18.本发明还涉及所述纳米相分离的固体聚合物电解质薄膜在锂金属电池固态电解质材料的应用。
19.进一步地,所述的电池为磷酸铁锂-石墨电池,磷酸铁锂-锂金属电池或锂三元过渡金属氧化物-锂金属电池。
20.本发明提供了一种纳米相分离的固体聚合物电解质薄膜及其制备方法和应用,其针对现有聚氧乙烯基醚的特点和提高室温离子电导率需求,结合聚苯乙烯-马来酸酐无规聚合物高机械强度、高热稳定性和小分子量聚乙二醇高锂离子电导率的优点,通过聚合物纳米相分离的结构设计,旨在解决现有聚合物固体电解质机械性能和室温离子电导率相互矛盾,高电压不稳定的问题。
21.本发明具有以下有益效果:
22.本发明所述聚合物固体电解质兼具高室温锂离子电导率、优异机械性能与宽电化学窗口,能够促进锂离子在锂负极均匀沉积并可以与高电压正极搭配,组装的固态锂金属电池和固态锂离子电池均得到较好的电池性能。
23.本发明通过调节主链苯乙烯-马来酸酐的比例以及换用带有更多苯环的单体来调节固态聚合物电解质的机械强度。聚合物固体电解质本身形成的纳米相分离可以提供宽的电化学窗口以及室温下高的离子电导率,另外聚合物电解质分子本身苯环间作用力的存在所产生的螺旋结构也会导致高的离子电导率。最后将制备的兼具高室温离子电导率与宽电化学窗口的固态聚合物电解质前驱体溶液流延到ptfe板里,利用溶液浇铸法成膜,可与不同正负极搭配组装成高性能固体电池。
附图说明
24.图1为本发明实施例1所制备的r-sma-g-peg的xrd图。
25.图2为本发明实施例2所制备的r-sma的电镜扫描图。
26.图3为本发明实施例3所制备的r-sma与r-sma-g-peg的红外对比图。
27.图4为本发明实施例1所制备得到的固态聚合物电解质在室温下与peg电解质的偏光显微镜照片对比。
28.图5为本发明实施例1所制备得到的固态聚合物电解质离子电导率随温度变化曲线图。
29.图6为本发明实施例1和实施例4所制备得固态聚合物电解质的电化学窗口对比图。
30.图7为本发明实施例1所制得的固态聚合物电解质的室温循环性能图。
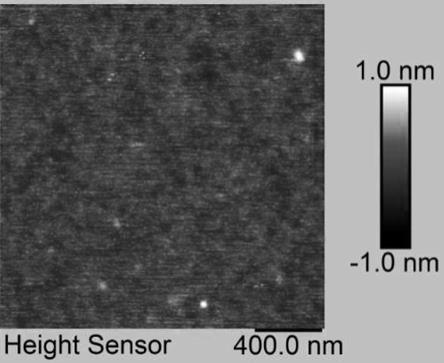
31.图8为本发明实施例1应用于钴酸锂电池的室温循环性能图。
32.图9为本发明实施例1与ncm811正极材料组装的电池在室温下的充放电曲线图。
33.图10为本发明实施例1、2、3、5合成的固态聚合物电解质的长循环性能对比图。
34.图11为本发明实施例1在锂硫电池中的循环性能图(其中硫负载量为1.5mgcm-2
)。
35.图12为实施例1制备的固态聚合物电解质的原子力显微镜图片。
具体实施方式
36.下面结合实施例,进一步阐明本发明,但不应理解为对本发明的限定。
37.实施例1:
38.在配有搅拌器,温度计,球形回流冷凝器和氮气保护装置的三口瓶中,加入10.34ml苯乙烯,然后将体系抽至真空并通入n2。0.981g马来酸酐和0.035g过氧化二异丙苯(dcp)溶于20ml碳酸二甲酯中超声分散均匀,之后转入恒压滴液漏斗。将油浴温度调至120℃,打开恒压滴液漏斗控制6s一滴,滴加完毕继续120℃恒温反应2h。反应液用大量乙醇溶液沉淀,得白色胶状物,用旋转蒸发仪旋去多余的溶剂,将产物90℃真空干燥9h,得产品r-sma,分子量约为6000。
39.在带有温度计、电动搅拌装置的四口烧瓶中,将共聚物r-sma,聚乙二醇(分子量为2000)和催化剂对甲苯磺酸水合物(tsoh.h2o)均溶于适量的四氢呋喃(thf)。在n2保护下加热至回流温度反应8h。冷却后用氢氧化钠乙醇溶液中和至ph=7,用旋转蒸发仪除去溶剂四氢呋喃和乙醇,然后真空干燥箱50℃干燥12h,得到产品r-sma-g-peg2000。其中sma与peg的质量比为1:2,催化剂的质量百分数为5%。所制备的r-sma-g-peg的xrd图见图1,120以及112处特征峰的出现证明peg被成功接枝到了r-sma上面。
40.将2g的r-sma-g-peg200080℃加热溶解于12ml的乙腈中,之后加入复配锂盐,复配锂盐为litfsi和lidfob摩尔比1:1;搅拌均匀,配置成聚合物前驱体溶液备用。将该溶液静置过夜然后进行超声脱泡处理,将前驱体溶液流延到ptfe板中放置于通风橱至溶液表面没有明显流动迹象,然后放入80℃真空干燥箱过夜干燥即得固态聚合物电解质。
41.得到的固态聚合物电解质在室温下与peg电解质的偏光显微镜照片对比如图4所示peg电解质在室温下出现了明显的黑十字消光结构且晶体很大,而本实施例制备的固态聚合物电解质在室温下没有结晶的出现,说明电解质内部无定形区域的增加,有利于锂离子的传输。图12为固态聚合物电解质1的原子力显微镜图片,可以看到有明显的相分离存在。相分离的存在可以打乱聚合物内部分子的规整排列以及提高电解质内部无定形区域的比例。
42.实施例2:
43.在配有搅拌器,温度计,球形回流冷凝器和氮气保护装置的三口瓶中,加入10.34ml苯乙烯,然后将体系抽至真空并通入n2。0.981g马来酸酐和0.035g过氧化二异丙苯(dcp)溶于40ml碳酸二甲酯中超声分散均匀,之后转入恒压滴液漏斗。将油浴温度调至120℃,打开恒压滴液漏斗控制6s一滴,滴加完毕继续120℃恒温反应2h。反应液用大量乙醇溶液沉淀,得白色胶状物,用旋转蒸发仪旋去多余的溶剂,将产物90℃真空干燥9h,得产品r-sma,分子量约为18000。
44.在带有温度计、电动搅拌装置的四口烧瓶中,将共聚物r-sma,聚乙二醇(分子量为2000)和催化剂对甲苯磺酸水合物(tsoh.h2o)均溶于适量的四氢呋喃(thf)。在n2保护下加热至回流温度反应8h。冷却后用氢氧化钠乙醇溶液中和至ph=7,用旋转蒸发仪除去溶剂四氢呋喃和乙醇,然后真空干燥箱50℃干燥12h,得到产品r-sma-g-peg2000。其中r-sma与peg的质量比为1:2,催化剂的质量百分数为5%。
45.将2g的r-sma-g-peg200080℃加热溶解于12ml的乙腈中,之后加复配锂盐,复配锂盐为litfsi和lidfob摩尔比1:1;搅拌均匀,配置成聚合物前驱体溶液备用。将该溶液静置
过夜然后进行超声脱泡处理,将前驱体溶液流延到ptfe板中放置于通风橱至溶液表面没有明显流动迹象,然后放入80℃真空干燥箱过夜干燥即得固态聚合物电解质2。图2为本发明实施例2所制备的r-sma的电镜扫描图,图2证明合成的r-sma为串珠状结构。
46.实施例3:
47.在配有搅拌器,温度计,球形回流冷凝器和氮气保护装置的三口瓶中,加入10.34ml苯乙烯,然后将体系抽至真空并通入n2。0.981g马来酸酐和0.035g过氧化二异丙苯(dcp)溶于20ml碳酸二甲酯中超声分散均匀,之后转入恒压滴液漏斗。将油浴温度调至120℃,打开恒压滴液漏斗控制6s一滴,滴加完毕继续120℃恒温反应2h。反应液用大量乙醇溶液沉淀,得白色胶状物,用旋转蒸发仪旋去多余的溶剂,将产物90℃真空干燥9h,得产品r-sma,分子量约为6000。
48.在带有温度计、电动搅拌装置的四口烧瓶中,将共聚物r-sma,聚乙二醇(分子量为6000)和催化剂对甲苯磺酸水合物(tsoh.h2o)均溶于适量的四氢呋喃(thf)。在n2保护下加热至回流温度反应8h。冷却后用氢氧化钠乙醇溶液中和至ph=7,用旋转蒸发仪除去溶剂四氢呋喃和乙醇,然后真空干燥箱50℃干燥12h,得到产品r-sma-g-peg6000。其中sma与peg的质量比为1:2,催化剂的质量百分数为5%。
49.将2g的r-sma-g-peg6000 80℃加热溶解于12ml的乙腈中,之后加复配锂盐,复配锂盐为litfsi和lidfob摩尔比1:1;搅拌均匀,配置成聚合物前驱体溶液备用。将该溶液静置过夜然后进行超声脱泡处理,将前驱体溶液流延到ptfe板中放置于通风橱至溶液表面没有明显流动迹象,然后放入80℃真空干燥箱过夜干燥即得固态聚合物电解质3。图3为本发明实施例3所制备的r-sma与r-sma-g-peg的红外对比图。与r-sma相比,r-sma-g-peg曲线中1856、1777cm-1
处c=o的消失以及1113和3417cm-1
出c-o-c的出现,都可以证明r-sma以及r-sma-g-peg的成功合成。
50.实施例4:利用实施例1的方法制备固态聚合物电解质膜4,区别仅在于,用lifsi代替复配锂盐。图6中为电解质膜4的电化学窗口,可以看到其在4v左右就发生了分解。
51.实施例5:利用实施例1的方法制备固态聚合物电解质膜5,区别仅在于,苯乙烯和马来酸酐的摩尔比为15:1,其中苯乙烯的质量为0.556g,马来酸酐的质量为0.035g。
52.实施例6:
53.将制备好的固态电解质膜1、2、3、5应用于固态锂电池,测试其不同温度下的离子电导率、电化学窗口、以及室温下磷酸铁锂电池的循环性能,详见图5、6、7、10。图5证明离子电导率随温度的变化,且离子电导率在25℃时可大于10-4
s/cm-1
。图6证明所述的固态电解质在室温下分解电压大于5v。图7证明,在25℃,0.2c的倍率下,循环300圈后放电容量还有121.3mah g-1
,容量保持率高达86.8%,平均库伦效率高达97%。图10表示不同实施例合成的固态聚合物电解质膜在25℃,0.5c倍率下长循环的性能图,其中固态聚合物电解质膜1的长循环性能最好,1000圈循环后还有40mah g-1
的容量,而固态聚合物电解质膜2、固态聚合物电解质膜3以及固态聚合物电解质膜5在300圈左右就已经发生短路
54.实施例7:
55.将制备好的固态电解质膜1与高压正极材料构建高能量密度锂金属电池。其中用固态电解质膜1代替隔膜,以金属锂作负极,钴酸锂和高压三元正极材料ncm811作正极,组装成电池测试其电化学性能。依次见图8、9。图8证明:在25℃,0.5c条件下循环100圈后放电
容量还有133mah g-1
,库伦效率达到90%。
56.图9表示所述的固态聚合物电解1质与高压正极材料匹配的充放电曲线,放电容量可达142mah g-1
。
57.实施例8:
58.将制备好的固态电解质膜1应用于锂硫电池,以金属锂作负极,多孔碳包覆的硫单质作正极,组装电池测试其室温下的循环性能。见图11。该电池具有良好的充放电性能,在约600mah g-1
条件下充放电250次。
59.上述实施例仅用来说明本发明的方法、详细工艺设备和工艺流程,但本发明并不局限于上述详细的工艺设备和工艺流程,即不意味着本发明必须依赖上述详细工艺设备和工艺流程才能实施。所属技术领域的技术人员应该明了,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以权利要求的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1