放射性核素锰-54的制备方法与流程
本发明涉及核燃料技术领域,尤其涉及一种放射性核素锰-54的制备方法。
背景技术:
国内尚未有54mn核素制备相关的公开发表物,国际上关于加速器制备54mn核素的报道也不多。
在加速器制备54mn的核反应及靶件选择方面,rwinj.等人报道了通过用质子辐照天然cr、富集54cr(80%)及氘核辐照天然fe进行加速器制备54mn的工作,核反应为54cr(p,n)54mn,56fe(d,α)54mn;h.t.russell报道了利用质子辐照富集54cr(65%)制备54mn的工作;p.m.smith-jones等人报道了利用富集109ag和56fe复合靶同时制备109cd、57co和54mn,54mn制备的核反应为56fe(d,α)54mn。靶核元素cr、fe的天然同位素组成中,cr的天然同位素组成为:50cr,4.345%;52cr,83.789%;53cr,9.501%;54cr(目标靶核),2.365%。fe的天然同位素组成为:54fe,5.845%;56fe(目标靶核),91.754%;57fe,2.119%;58fe,0.282%。
上述靶件选择均存在下述问题:富集靶价格昂贵,经济性不高;天然靶件中,目标靶核元素54cr、56fe在其天然同位素组成中占比较低且非靶核元素组成复杂,导致使用cr或者fe的同位素作为靶核进行核反应时将存在较多的副反应而生成大量放射性杂质核素,使放化分离流程复杂化。
klosmalgorzata等人的论文《productionandseparationofmanganese-54fromalpha-irradiatedv2o5target》中,提出用20mev的α粒子辐照v2o5靶制备得到核纯度为99.9%的54mn,但是其采用的v2o5传热性能较差,需要与al等导热性较强的金属衬底连接使用。但是,上述方法靶件制备繁琐且能承受的辐照束流较小,存在熔靶风险且限制54mn产额,金属衬底可能反应生成放射性杂质从而增加放化分离难度;其束流选用20mev的α粒子,反应截面低,且会通过50v(α,2n)52mn、51v(α,2n)53mn反应产生更多的放射性杂质,需要通过两个月左右时间进行“冷却”以去除短寿命核素52mn、54mn,制备周期长。
c.s.sastri等人的论文《productionofmanganese-52ofhighisotopicpurityby3he-activationofvanadium》中,提出从辐照金属钒靶与其他放射性杂质(51cr、48/49v)中分离提纯52mn(t1/2=5.59d)的方法,52mn回收率为50-60%,核纯度大于99.9%。上述方法采用浓硝酸溶解v箔,溶靶过程反应激烈、操作安全性不高,且溶靶过程中加入试剂较多且需加热煮沸操作,溶靶过程繁琐且不经济;放化分离流程中使用8-羟基喹啉有机相进行52mn萃取,操作繁琐,易产生气溶胶,造成放射性污染,有机废液处难度大且52mn回收率低。
技术实现要素:
本发明要解决的技术问题在于,提供一种经济、操作简单且安全性高的放射性核素锰-54的制备方法。
本发明解决其技术问题所采用的技术方案是:提供一种放射性核素锰-54的制备方法,包括以下步骤:
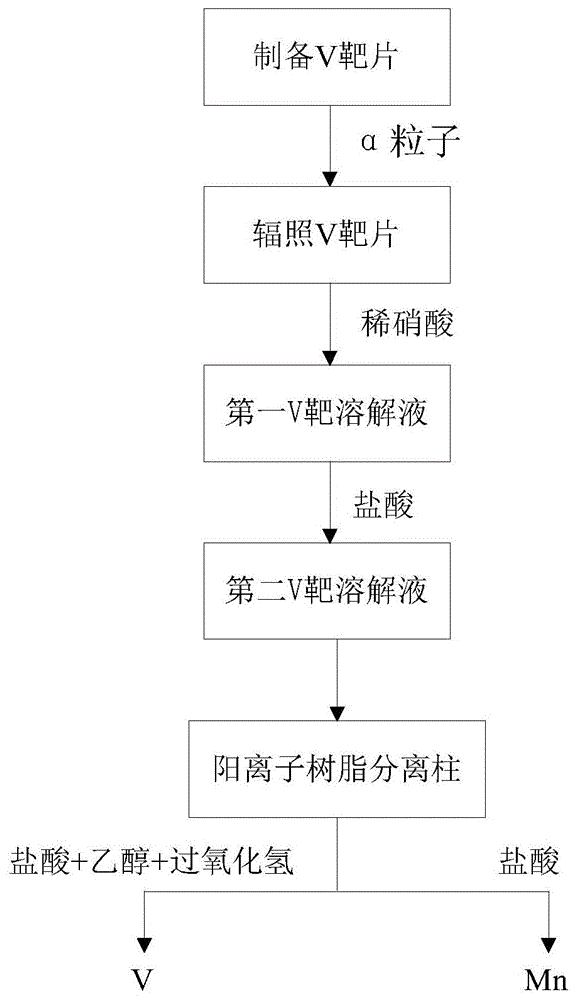
s1、制备v靶;
s2、采用10mev-12mev的α粒子辐照v靶;
s3、将辐照后v靶溶解于稀硝酸中,形成第一v靶溶解液;
s4、将所述v靶溶解液蒸干后再溶解于盐酸中形成第二v靶溶解液;
s5、将所述第二v靶溶解液加载到阳离子树脂分离柱中,采用由盐酸、乙醇和过氧化氢形成的混合溶液进行淋洗;
s6、用浓度为2mol/l-5mol/l的盐酸溶液对所述阳离子树脂分离柱中的mn进行解吸处理,得到含有54mn的mncl2溶液。
优选地,步骤s1中,所述v靶为天然v靶或者富集51v靶。
优选地,所述v靶为天然v靶制成的v靶片。
优选地,步骤s3中,所述稀硝酸的浓度为3mol/l-8mol/l。
优选地,步骤s3中,将所述稀硝酸以多次添加的方式对v靶片进行溶解;在添加一次稀硝酸静置反应后,吸取转移反应后的溶解液后再进行下一次稀硝酸的添加。
优选地,步骤s4中,所述盐酸的浓度为0.1mol/l-0.5mol/l。
优选地,步骤s5中,所述混合溶液中,盐酸和乙醇的体积比为5:1,过氧化氢在混合溶液中的体积占比为0.1-1%;该盐酸的浓度为0.1mol/l-0.5mol/l;所述过氧化氢的浓度为3-5%。
优选地,步骤s5中,淋洗的流速为0.3ml/min-0.6ml/min。
优选地,步骤s5中,阳离子树脂分离柱的树脂粒径为50-100目,预先依次经过去离子水和浓度0.1mol/l-0.5mol/l的hcl浸泡。
优选地,步骤s6中,解吸处理以解吸、浸泡依次循环多次进行。
优选地,在所述步骤s2中,辐照束流强度为10-100μa,粒子入射角度1°-10°,辐照时间6-18h。
本发明的放射性核素锰-54(54mn)的制备方法,基于51v(α,n)54mn核反应进行54mn的制备,采用稀硝酸进行v靶溶解,反应温和,操作简单且安全性高;选择特定的入射α粒子能量范围,在保障较高反应截面的基础上,有效避免了多余的副反应放射性产物生成,无需“冷却”时间,制备周期短,制备产额高。
另外,选用天然v靶作为靶件,天然同位素组成简单、所需靶核素天然丰度高,一方面,核反应副反应少,进而简化后续放化分离操作;另一方面,极高的靶核素天然丰度降低了富集靶使用的必要性,进而降低54mn制备成本,具有靶件制备简单经济、放射性杂质副产物少、放化分离操作简单、废液料少且易处理、54mn回收率及放射性核纯度高的优点。
附图说明
下面将结合附图及实施例对本发明作进一步说明,附图中:
图1是本发明一实施例的放射性核素锰-54的制备方法的流程示意图;
图2是本发明一实施例制得的含54mn的mncl2溶液的γ能谱图。
具体实施方式
参考图1,本发明一实施例的放射性核素锰-54(54mn)的制备方法,可包括以下步骤:
s1、制备v靶。
v靶可以是天然v靶或者富集51v靶。
天然v靶或者富集51v靶可以金属形式或氧化物形式存在。
v(钒)可通过电镀等形式附着在靶件衬底上,也可直接通过v板按照加速器靶托尺寸加工形成。由于富集51v靶价格昂贵,优选采用天然v靶,更具经济性。
由于氧化物传热性较差,难以承受高束流强度的粒子轰击,金属v靶相较于v2o5靶具备更优异的导热性能和辐照安全性,因此优选以金属形式存在的天然v靶。具体地,采用v板按照加速器靶托尺寸加工形成v靶片。
s2、采用10mev-12mev的α粒子辐照v靶。
v靶片的厚度大于α粒子的射程(37.5μm)。在本实施例中,辐照束流强度为10-100μa,粒子入射角度1°-10°,辐照时间6-18h。
具体地,天然v靶的同位素组成如下:50v,0.250%;51v,99.750%。通常,用α粒子照射天然v靶主要发生(α,n)、(α,2n)、(α,d)、(α,p)等核反应,主要核反应及生成产物如下表1所示。
表1
为了得到放射性核纯度大于99.9%的54mn溶液,需要通过辐照离子能量选取以及放化分离流程设计去除辐照天然v靶中规定v、cr等非放核素以及52mn、53mn等放射性杂质。
其中,51v(α,n)54mn反应阈能为2.5mev并在12mev左右反应截面最高,50v(α,2n)52mn及50v(α,n)53mn的反应阈能分别为13.2mev、12.1mev,因此控制入射α粒子能量在8mev-12mev时即可避免50v(α,2n)52mn及50v(α,n)53mn反应发生,且能保障具有较高反应截面。虽然仍会经50v(α,n)53mn反应产生53mn,但是一方面50v仅占靶件含量的0.250%,且反应截面小,53mn产量可忽略;另一方面,53mn半衰期t1/2=3.7×106a,且不发射γ射线,与目标产物54mn相比,可被视为稳定核素。进一步地,入射α粒子能量控制在10mev-12mev之间,能够较稳妥得避免因入射离子能量歧离导致上述副反应,且对最大化产额最有利。
s3、将辐照后v靶溶解于稀硝酸中,形成第一v靶溶解液。
稀硝酸的浓度为3mol/l-8mol/l,相较于浓硝酸反应更温和且溶靶安全性更高。并且,在上述浓度范围内,反应速度随硝酸浓度增大而增大,因此8mol/lhno3更利于v靶溶解。
用hno3溶解辐照v靶时,将稀硝酸以多次添加的方式对v靶片进行溶解;在添加一次稀硝酸静置反应后,吸取转移反应后的溶解液后再进行下一次稀硝酸的添加。即:采用少量多次溶解方式,每次添加0.5-1mlhno3静置反应10-20min后进行溶解液吸取转移,通过多次滴加吸取得到所有辐照v靶溶解液(即第一v靶溶解液)。
为确保溶解完全,可通过伽马辐射探测仪对溶解后剩余靶进行放射性检测以判定放射性产物是否溶解完全。
s4、将第一v靶溶解液蒸干后再溶解于盐酸中形成第二v靶溶解液。
盐酸的浓度为0.1mol/l-0.5mol/l。
s5、将第二v靶溶解液加载到阳离子树脂分离柱中,采用由盐酸、乙醇和过氧化氢形成的混合溶液进行淋洗,直至淋洗液中的v含量为零。
混合溶液中,盐酸和乙醇的体积比为5:1,过氧化氢在混合溶液中的体积占比为0.1-1%。其中,盐酸的浓度为0.1mol/l-0.5mol/l;过氧化氢的浓度为3-5%。
淋洗时,控制淋洗的流速为0.3ml/min-0.6ml/min。
其中,阳离子树脂分离柱采用粒径为50-100目的树脂,树脂预先经过去离子水浸泡24-36h,再经过浓度0.1mol/l-0.5mol/l的hcl浸泡数个小时。
作为选择,阳离子树脂分离柱采用dowex-50×8阳离子树脂分离柱。
采用dowex-50×8阳离子树脂分离柱时,预先将阳离子分离柱用去离子水冲洗;取dowex-50×8树脂,清洗后烘干,用研磨机研磨,再用分子筛分筛,取约1.5g50-100目范围的dowex-50×8树脂,先用200ml去离子水浸泡24h,再用100ml0.2mol/lhcl浸泡6小时,之后将树脂上阳离子分离柱。阳离子分离柱的玻璃柱底部、树脂顶部均使用玻璃纤维盖住以确保树脂不被淋洗液冲起。
s6、用浓度为2mol/l-5mol/l的盐酸溶液对阳离子树脂分离柱中的mn进行解吸处理,直至解吸完全,得到含有54mn的mncl2溶液。
优选地,解吸处理以解吸、浸泡依次循环多次进行,即:解吸、浸泡、再解吸、再浸泡的多次重复操作。
图2为本发明制得的mncl2溶液的伽马能谱图,从图中可以看出,能谱区只有54mn的特征能谱(834.8kev),无其它核素的能谱,54mn的放射性核纯度达99.9%,总的放射性活度约为252μci,说明得到了高纯度的无载体[54mn]mncl2溶液。
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的等效结构或等效流程变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!