作为具有形态发生活性的包被层和支架的非晶形无机聚磷酸盐‑磷酸钙和碳酸钙颗粒的制作方法
本发明涉及制备基于无机聚磷酸盐(聚p)和磷酸钙或碳酸钙的显示成骨活性的非晶形、纳米颗粒材料或微粒材料的方法。在本发明的一个方面,本发明人显示非晶形聚磷酸钙(ca-聚p)微粒可用于钛合金表面的生物功能化。本发明的方法允许在钛氧化的ti-6al-4v支架上制造耐久且稳定的、几乎均匀且具有形态发生活性的ca-聚p层,与生物惰性的非改性钛表面相比,其支持骨细胞的生长并增强骨细胞的功能活性。优选的方面涉及在低浓度聚磷酸钠(na-聚p)的存在下形成非晶形磷酸钙(cap)颗粒。这种材料导致参与骨形成的蛋白的表达强烈上调。本发明的另一方面涉及含有聚p稳定的acc和少量球霰石的材料,该材料在动物实验中显示成骨活性并支持植入区域的再生。本发明的非晶形材料具有用于骨植入物的潜力。
背景技术:
除了胶原蛋白和水之外,骨骼的基本构成块还包含碳酸化磷灰石[ca5(po4,co3)3(oh)]以及羟基磷灰石(ha)。结晶矿物质可能由非晶形磷酸钙(acp)形成(wangy,etal.(2013)water-mediatedstructuringofboneapatite.natmater12:1144-1153)。
最近的证据表明非晶形碳酸钙(acc)作为用于形成acp和碳酸化磷灰石的生物种子,其是由碳酸酐酶(ca),很可能是由可溶性ca-ii同种型和/或细胞膜相关ca-ix形成的材料(wangxh,etal.(2014)modulationoftheinitialmineralizationprocessofsaos-2cellsbycarbonicanhydraseactivatorsandpolyphoshate.calciftissueint94:495-509;müllerweg,etal.mineralizationofbone-relatedsaos-2cellsunderphysiologicalhypoxicconditions.febsj.2015oct10)。
acc是碳酸钙最不稳定的多晶型物,其存在于非晶形相和结晶相中;在三种主要的结晶多晶型物球霰石、霰石和方解石中,亚稳态球霰石是结晶caco3的热力学最不稳定形式(meldrumfc,
因此,可以将骨ha的形成细分为以下三个机械上不同的阶段:
1.通过碳酸酐酶来酶促形成acc生物种子;
2.在acp形成下,通过磷酸盐来非酶促交换碳酸盐离子;和
3.将acp转化为结晶相碳酸化磷灰石/ha。
acc作为潜在的诱导再生/支撑材料的应用,受到acc本身不稳定这一事实的阻碍。acc在体内的稳定性通过通常与mg2+结合的专门蛋白调节,而在体外条件下,非生物来源添加剂如可溶性聚羧酸盐,还有mg2+、三磷酸盐或聚磷酸盐种类,将acc冻结到相对稳定的相(kellermeierm,etal.(2010)stabilizationofamorphouscalciumcarbonateininorganicsilica-richenvironments.jamchemsoc132:17859-17866)。
相反,球霰石足够稳定以允许解离,反过来又可以作为骨再生的潜在离子缓冲系统,并且由此可以修改caco3向ha的转化过程(
最近,本发明人成功地制备了基于非晶形聚磷酸盐(聚p)的微粒(müllerweg,etal.anewpolyphoshatecalciummaterialwithmorphogeneticactivity.materialsletters2015;148:163-166;müllerweg,etal.retinolencapsulatedintoamorphousca2+polyphoshatenanospheresactssynergisticallyinmc3t3-elcells.eurjpharmbiopharm2015;93:214-223),该微粒不仅在成骨细胞系统上以合成代谢方式起作用,而且还为它们的靶细胞(如成骨细胞)提供代谢燃料,并引起细胞内atp水平提高以及线粒体数量的增加(müllerweg,etal.amorphousca2+polyphoshatenanoparticlesregulatetheatplevelinbone-likesaos-2cells.jcellsci2015;128:2202-2207)。本发明人还在gb1420363.2中公开了这种生物相容性的、生物可降解的和具有生物活性的基于聚p的材料。在gb1420363.2中描述的微粒的大小,可以通过确定的pi:ca2+摩尔比来调节(müllerweg,etal.(2015)anewpolyphoshatecalciummaterialwithmorphogeneticactivity.materialslett148:163-166)。所形成的颗粒保持非晶形状态,因此容易被碱性磷酸酶(alp)酶促水解。
聚p在血液中以相当大的量存在并且较大程度存在血小板中,并且已经被认为是用于形成骨磷酸钙沉积物的磷酸盐来源。通过alp从该聚合物酶促去除正磷酸盐,其可以用作骨矿化的供体。此前,发明人描述了聚p调节细胞特异性分化过程,例如用模型细胞系saos-2细胞将矿物沉积物形成到骨形成成骨细胞上,使用模型细胞系raw264.7,诱导alp以及使opg(骨保护素):rankl(核因子κb配体的受体激活剂)的比例向合成代谢的成骨细胞途径改变,并由此抑制破骨细胞的功能。此外,聚p诱导用于编码骨形态发生蛋白-2(bmp-2)和支架结构纤维所成的系统胶原蛋白的基因。酶介导的骨形成中的现有技术和聚p的作用已被描述于,例如:
wangxh,
wangxh,etal.(2015)polyphoshateasametabolicfuelinmetazoa:afoundationalbreakthroughinventionforbiomedicalapplications.biotechnolj.doi:10.1002/biot.201500168;
müllerweg,etal.(2015)non-enzymatictransformationofamorphouscaco3intocalciumphosphatemineralafterexposuretosodiumphosphateinvitro:implicationsforinvivohydroxyapatiteboneformation.chembiochem16:1323-1332;和
müllerweg,etal.polyphoshate:amorphogeneticallyactiveimplantmaterialservingasmetabolicfuelforboneregeneration.macromolecbiosci2015;15:1182-1197。
因为现有材料的局限性(例如患者的长期固定期,植入物引起的感染等),迫切需要新的骨植入材料。理想地,这些植入物必须遵循骨形成的自然过程的原理,允许受损骨组织的快速再生。
在本发明的一个方面,发明人描述了聚p可以稳定acc相。在本发明的方法中,在5%[w/w]的水平下,聚p显著抑制acc转化为结晶caco3,在10%[w/w]的百分比下,该聚合物几乎完全阻断了该过程。这个发现是意想不到的。以前已经报道,可以将掺有规定ca2+摩尔比的可溶性na-聚p加工成固体纳米尺度的纳米颗粒/微粒,其保持非晶形(müllerweg,etal.(2015)anewpolyphosphatecalciummaterialwithmorphogeneticactivity.materialsletters148:163-166;müllerweg,etal.(2015)retinolencapsulatedintoamorphousca2+nanospheresactssynergisticallyinmc3t3-e1cells.eurjpharmbiopharm93:214-223)。不能期望,ca-聚p可以作为亚稳态acc的稳定剂;另参见:gb1420363.2和gb1502116.5。
聚p作为骨细胞上的具有形态发生活性的无机分子,诱导其矿化(gb1420363.2、gb1502116.5、gb1403899.6)。在本申请中,发明人还显示含有5%[w/w]或10%[w/w]聚p的caco3,在saos-2细胞以及人间充质干细胞(msc)中通过诱导alp和骨形态发生蛋白2(bmp2)的基因表达而包含成骨潜能。更令人惊奇和意外的是,acc以“协同”方式增强了聚p对bmp2表达的刺激作用。此外,本发明人证明,本发明的acc/聚p混合材料是生物相容性的并且在体内支持再生,使其成为用于骨替换/植入物的有希望的支架材料。
在本发明的另一方面,本发明人使用用于从氯化钙和磷酸氢二铵开始制备cap的技术。在新颖的方法中,除了用10:6的特征性ca/p摩尔比制备ha之外,他们还制备了混合有各种量聚p的cap。发明人意外地发现,含有>10重量%聚p的cap制品(相对于改性cap/ha沉积物)是非晶形的,而含有<10重量%聚p的cap/ha样品由结晶相组成。发现所有样品都支持骨细胞相关saos-2细胞的生长,但令人惊奇的是,只有含有10重量%(wt.%)的聚p的cap制品引起强的形态发生活性,如通过测量用于编码alp和i型胶原蛋白的基因(骨和骨相关细胞分化的标记基因)表达所确定的。基于这些结果,本发明的基于聚p/cap的材料可能有益于作为骨替代植入物的应用。
此前,本发明人开发了在规定浓度cacl2存在下于环境条件下制备的基于聚p的材料。该材料由生物相容性的和生物可降解的球形非晶形颗粒组成。
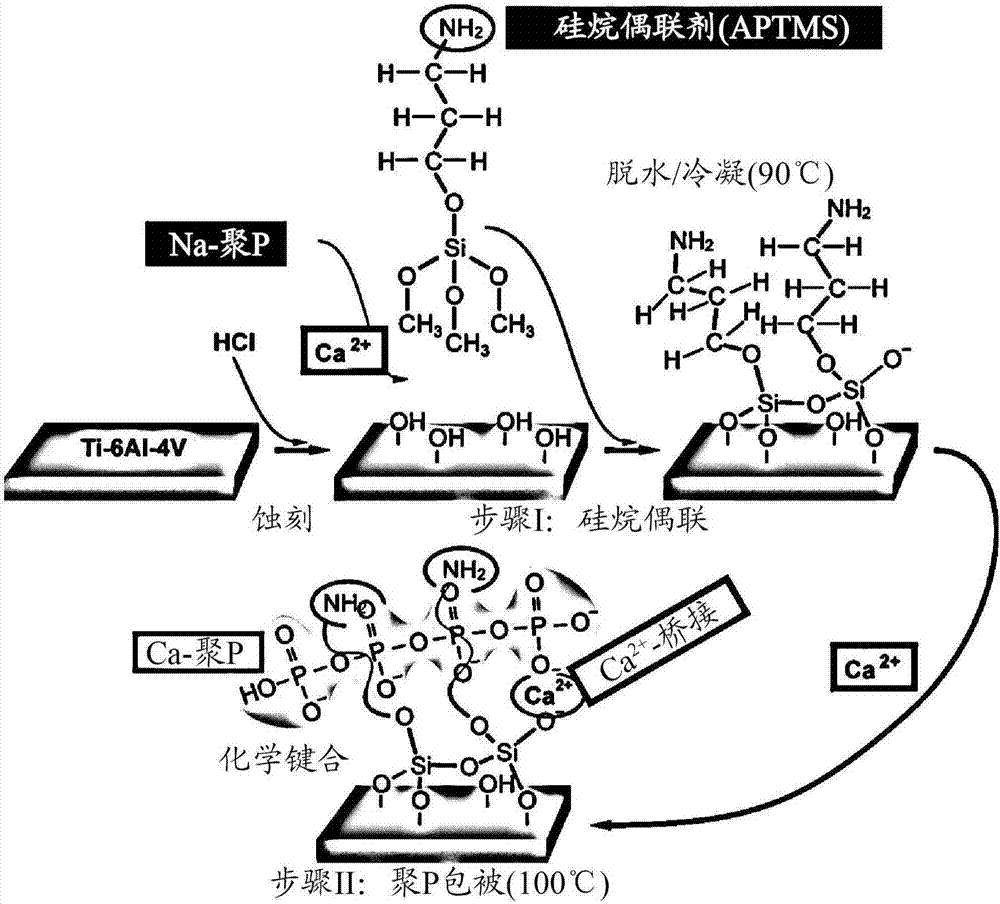
现在发明人惊奇地发现,可将这种以微粒范围尺寸制备的具有生物活性的材料用于通过形成到硅烷偶联剂aptms的ca2+离子桥接的ti-6a1-4v支架的表面包被层,如电子显微镜和元素分析(edx)方法所证明。该发现和包被层的高耐久性和稳定性是出乎意料的,尤其是由于ca-聚p颗粒的微粒性质。
ca-聚p包被的钛合金的另一令人惊奇的特性是,其作为骨样saos-2细胞生长的合适基底的特性,尽管其明显降低的表面粗糙度不可能支持细胞附着,甚至更多,与未处理的钛支架相比,其诱导这些细胞表达关键酶(碳酸酐酶ix(caix)和alp)的能力,所述酶参与启动酶诱导的骨矿物质沉积。
基于它们的特性,本发明的包被的钛支架是用于制备具有新颖的再生诱发特征的高精度植入物的有希望的材料,其可以以个体化的尺寸和形状生产。
众所周知,na-聚p容易螯合ca2+并形成不溶性沉淀。因此,可以预期,向cacl2和(nh4)2hpo4中加入na-聚p将导致非晶形ca-聚p和结晶cap(ha)矿物沉积物的共沉淀。
在本发明的另一方面,本发明人在沉淀过程中,与cacl2和(nh4)2hpo4(用于在水溶液中形成ha的底物)一起加入了增加浓度的na-聚p。令人惊奇和意外的是,他们发现,在同时制备尺寸约为100nm的非晶形聚p/cap混合颗粒的情况下,10wt.%的聚p的含量防止了结晶ha的形成。以本发明这个方面为基础的结果总结如图1所示。
这一发现是重要的,因为由于不利的物理(低溶解度)和机械特性(脆性),即使是生物相容性的结晶ha,其应用目前也仅限于粉末、包被层和多孔体以及非承重植入物(wangmc,etal.crystallinesize,microstructureandbiocompatibilityofhydroxyapatitenanopowdersbyhydrolysisofcalciumhydrogenphosphatedehydrate(dcpd).ceramicsintern2015;41:2999-3008)。如果cap可以在非晶形相中合成,则可以避免这些缺点。
本发明人证明,非晶形聚p/cap颗粒(缩写:acap-聚p)结合细胞生长促进活性显示出不同的形态发生活性。他们发现acap-聚p引起两个用于骨形成的标记基因(i型胶原蛋白和alp)的强烈上调。acap-聚p的效力与ca-聚p相当。
基于其引发形态发生活性的特性,本发明的acap-聚p颗粒提供了用作人造骨植入物的有希望的材料,其由生理代谢物/聚合物制备。
在本发明的另一方面,本发明人惊奇地发现,可通过聚p来稳定亚稳态acc相。在人骨中,acc形成为结晶碳酸化磷灰石/ha的前体。聚p用作非酶促碳酸盐/磷酸盐交换的磷酸盐源。本发明人证明,关于caco3,在5%[w/w](称为“ccp5”)的百分比时,聚p抑制acc向结晶caco3转变,而在10%[w/w](称为“ccp10”)时几乎完全抑制。他们显示两种制品都是非晶形的,但也含有少量的球霰石,如通过xrd、ftir和sem分析所显示的。
本发明人证明,与方解石相反,本发明的acc/聚p颗粒显示成骨活性。它们诱导saos-2细胞以及人间充质干细胞(msc)中编码alp的基因的表达,以及bmp2基因的表达。此外,在大鼠的体内研究中,使用含有本发明的acc/聚p材料并插入动物背部肌肉的plga微球体,本发明人证明包封的acc/聚p颗粒不仅是生物相容性的,而且还支持植入区域的再生。令人惊奇的是,与生物惰性方解石相比,含有少量球霰石的acc具有成骨潜力和优异的特性。基于这些特性,本发明的材料代表用于骨植入物的有希望的支架材料。
以下关于聚p的专利申请被认为是相关的:gb1406840.7、gb1403899.6、wo2012/010520、gb1420363.2、gb1502116.5和gb1510772.5。
在gb1420363.2中,本发明人公开了制备由钙-聚p微粒组成的材料的方法,该材料显示出以下特性:(i)它是非晶形的,和(ii)在能够矿化的细胞系统中具有生物活性。
结果也报道于:müllerweg,etal.anewpolyphosphatecalciummaterialwithmorphogeneticactivity.materialsletters2015;148:163-166;müllerweg,etal.retinolencapsulatedintoamorphousca2+polyphosphatenanospheresactssynergisticallyinmc3t3-e1cells.eurjpharmbiopharm2015;93:214-223;和müllerweg,etal.polyphosphate:amorphogeneticallyactiveimplantmaterialservingasmetabolicfuelforboneregeneration.macromolecbiosci2015;15:1182-1197。
在gb1420363.2中描述的材料的特性,优于ha(也参见实施例)以及用于骨再生和修复的常规聚p制品(例如gb1406840.7和gb1403899.6)。
现在,本发明人成功地开发出通过其可以用聚p紧密地覆盖钛/钛合金的步骤。在用hcl蚀刻后,金属表面与aptms共价连接,之后ca-聚p颗粒可以通过ca2+离子键连接到所述表面(图2)。
在金属表面的本发明的聚p包被,证明是耐用的并且令人惊奇地稳定。
aptms可以被其它硅烷偶联剂代替,例如3-(三甲氧基硅基)丙基甲基丙烯酸酯。除了聚p之外,aptms另外的官能团允许肽与硅烷包被的钛表面结合。
与未处理的钛圆片相当高的表面粗糙度(最大约为7μm)相比,ti-ca-聚p圆片是平滑的,最大粗糙度为0.8μm。通常,与中等粗糙表面相比,细胞附着到非常光滑表面的程度较低(例如,huanghh,etal.effectofsurfaceroughnessofgroundtitaniumoninitialcelladhesion.biomoleng2004;21:93-97)。因此,出乎意料的是,在没有任何圆片的对照试验中看到,聚p包被的圆片允许saos-2细胞以一定速率生长。
发明人显示,在2天的孵育期后,在含有未处理的钛圆片试验中的细胞死亡。这与在这段时间内saos-2细胞密集地附着到ti-ca-聚p圆片并形成几乎均匀的单细胞层的观察结果形成鲜明对比。令人惊讶的发现是,在ti-ca-聚p圆片上生长的细胞显示细胞扩散的表型形态,这是活性细胞代谢和细胞迁移的明显标志。
“关于”表示所示值的+/-10%。
本发明的另一方面涉及以下发现:本发明的ca-聚p包被层能够刺激骨形成细胞的功能活性,如通过在包被的金属表面(与未处理的钛表面相比)上生长的细胞中编码碳酸酐酶ix(caix)和alp的转录物的增加的稳态水平所证明(如用qrt-pcr定量的)。
酶ca参与骨形成的引发(形成caco3沉积物;müllerweg,etal.inductionofcarbonicanhydraseinsaos-2cells,exposedtobicarbonateandconsequencesforcalciumphosphatecrystalformation.biomaterials2013;34:8671-8680;wangxh,etal.enzyme-basedbiosilicaandbiocalcite:biomaterialsforthefutureinregenerativemedicine.trendsbiotechnol2014;32:441-447)。
alp是具有功能活性的矿物沉积而形成成骨细胞的确定标志物(参见:wangxh,etal.bio-silicaandbio-polyphosphate:applicationsinbiomedicine(boneformation).curropinbiotechnol2012;23:570-578)。
本发明的数据显示钛氧化的ti-6a1-4v支架是用于体外骨样saos-2细胞的惰性基底。如果包被具有形态发生活性的ca-聚p聚合物,则该金属获得生物功能特性。具有聚p的这种植入材料的生物功能化的进展,不仅提供个体化植入物的制备,而且还提供了将硬而脆的金属植入物的机械特性与较软的骨骼及其周围组织的机械特性相匹配的有利特性。
聚p的链长可以在约3个至约1000个磷酸盐单元的范围,优选在约20个至约200个磷酸盐单元的范围,以及最优选约40个磷酸盐单元。
本发明方法中使用的ca-聚p微粒的优选组成是化学计量比(磷酸盐与钙)为:0.1比1至50比1,优选1比1至10比1,以及最优选7比1。
尽管它们的直径(0.2μm和3μm)在允许受体介导的胞吞作用(约50nm)的范围之外,出人意料的是,ca-聚p微粒是具有生物活性的。
与通过常规技术制备的ca-聚p盐相比,该聚p材料是生物可降解的,并且显示出优异的形态发生活性。
本发明方法的另一方面是,应用/使用该方法来制备具有生物活性的钛合金植入物。本发明方法的另一方面是,应用/使用该方法来制备刺激成骨细胞活性的植入物。本发明方法的另一方面是,将ca-聚p包被的钛合金表面和植入物与镓盐组合应用/使用,以便利用它们在通过应用本发明方法所制备的包被层上的增强的协同效应。发明人意外地发现,镓盐(如硝酸镓)增强了具有生物活性的ca-聚pti合金包被层对具有功能活性特征的成骨细胞的转录物的表达、稳态水平的刺激作用。这一发现令人惊奇,因为已经报道镓盐只能通过破骨细胞调节骨吸收,但不影响或仅最低限度影响成骨细胞的基因表达和alp活性(verrone,etal.galliummodulatesosteoclasticboneresorptioninvitrowithoutaffectingosteoblasts.brjpharmacol2010;159:1681-1692)。
本发明的另一方面涉及令人惊奇的发现,即如果在通常导致结晶ha合成的步骤中,聚p以一定浓度存在,也可以制备与结晶ha相比具有优异特性,并且实现与gb1420363.2中公开的聚p微粒几乎相同的生物活性(形态发生活性、刺激骨相关基因表达)的含有非晶形聚p的材料。
由本发明人开发的制备具有生物活性的非晶形聚p取代的cap颗粒(acap-聚p)的本发明方法,包括以下步骤:
a)将聚p盐的水溶液加入到磷酸盐源的水溶液中;
b)将所得溶液加入到溶解的钙盐中;
c)将ph调节至碱性值,优选约10;和
d)收集、洗涤和干燥数小时后,优选在室温下24小时后形成的所得沉淀。
聚p盐优选为聚磷酸钠(na-聚p)。如果聚p盐的量高于5wt.%的聚p盐,则形成本发明的聚p-取代的cap颗粒(acap-聚p),称为cap制品。作为实例,用10wt.%的聚p盐的聚p取代的cap颗粒(acap-聚p)已经实现了最佳结果。
根据本发明方法制备的形成本发明聚p取代的cap颗粒(acap-聚p)的cap成分的钙盐和磷酸盐源,可以分别是氯化钙(cacl2)和磷酸氢二铵[(nh4)2hpo4]]。
通过本发明方法制备的聚p取代的cap颗粒(acap-聚p)获得最佳结果,其中计算钙盐的量和用作磷酸盐源的试剂的量以获得ca/p摩尔比为10:6的cap。
聚p取代的cap颗粒(acap-聚p)的平均尺寸可以在约20nm至约300nm的范围,优选在约70nm至约120nm的尺寸范围。
本发明的另一方面涉及以下发现:本发明的聚p-取代的cap颗粒(acap-聚p)能够刺激骨形成细胞的功能活性,如通过在骨形成saos-2细胞中编码i型胶原蛋白(col-i)和碱性磷酸酶(alp)的转录物的增加的稳态水平所证明(如用qrt-pcr定量的)。
意外地是,这些聚p取代的cap颗粒(acap-聚p)具有生物活性,尽管它们的直径(70-120nm)大于可被受体介导的内吞作用(约50nm)摄取的颗粒的直径。
与通过常规技术制备的ha晶体相比,聚p取代的cap颗粒(acap-聚p)是生物可降解的,并且显示出优异的形态发生活性。
本发明方法的另一方面是,应用该方法制备生物活性植入材料。本发明方法的另一方面是,应用该方法来制备刺激成骨细胞活性的人造骨植入物。
本发明的另一方面涉及制备含有少量球霰石的acc多晶型物。发明人在合成acc期间,将阴离子聚合物聚p的na+盐加入到caco3(cacl2和na2co3)的前体中(图3)。令人惊奇的是,本发明人发现,在最终浓度为10%时聚p几乎完全防止acc转变为其结晶多晶型物球霰石、霰石和方解石。
分别测试的caco3固体和聚p生理代谢物都具有成骨潜力,并且可以作为具有生物活性的骨移植物的成分。反过来,所开发的支架不仅利用聚p的形态发生潜力,而且还利用该聚合物的特性在acc阶段冷冻caco3固体。该材料的成骨活性优于方解石;它通过刺激成骨细胞来强烈诱导编码alp(骨形成标志物)的基因的表达。该结果已经从骨样saos-2细胞和msc的研究中获得。
此外,发明人证明,acc/聚p通过成骨细胞强烈上调bmp2(骨形成诱导物)的表达。更重要的是:他们惊奇地发现acc以“协同”的方式增加由聚p诱导的bmp2表达,导致bmp2转录水平的更快上升。可以预期本发明的acc/聚p微粒的这种效应将导致骨缺损的更快愈合。
acc/聚p材料不仅具有生物相容性,而且还支持受损植入区域的细胞再生。为了评估acc/聚p材料在体内的生物相容性,本发明人将本发明的材料包封在plga微球体中。并行地,对照球体保持没有acc/聚p。将珍珠/微球嵌入大鼠背部的肌肉。观察期2周、4周和8周后,从大鼠中取出组织样本,并在切片和用mayer苏木精染色后进行显微镜检查。检查显示,在具有含有acc/聚p材料的微球的动物中,分别在4周和8周后,具有细胞的植入物区域的晚期增殖(repopulation)变得明显。相反,缺少acc/聚p的微球没有任何细胞。通过测量植入区域组织样本的硬度(中值redym刚度)来支持这些结果,其显示了在8周时间段后与对照相比显著增加1.8倍(植入前在肌肉样品中测量值的81%)。
本发明人开发的用于制备本发明的acc/聚p材料的优选方法,包括以下步骤:
a)在例如1升0.1mnaoh中制备含有聚p盐的溶液;
b)向该溶液中加入0.5mol/l的na2co3;
c)用1.5倍体积的去离子水稀释所得溶液;
d)将该溶液与相同体积的含有0.5mol/lcacl2·2h2o的水溶液快速混合(导致钙离子和碳酸根离子之间的等摩尔浓度比);和
e)在室温下用丙酮洗涤后,过滤并干燥沉淀物。
用于制备本发明的acc/聚p微粒的0.1mnaoh溶液中聚p盐的优选浓度,在0.001mol/l至1.0mol/l的范围,优选在0.01mol/l至0.1mol/l的范围(基于磷酸盐单位)。
如果用于制备本发明的acc/聚p微粒的0.1mnaoh溶液中的聚p盐的浓度为0.025mol/l,或者,甚至更优选为0.05mol/l(基于磷酸盐),则获得最佳结果。所得的制品分别称为“ccp5”和“ccp10”。聚p盐优选为na-聚p。
聚p的链长可以在3个至约1000个磷酸盐单元的范围。具有平均链长为约10个至约100个磷酸盐单元的聚p分子获得最佳结果,并且在该范围内最佳约40个磷酸盐单元。
本发明的另一方面涉及以下发现:本发明的acc/聚p颗粒通过在骨形成saos-2细胞中(如用qrt-pcr定量的)诱导编码alp和bmp2的基因表达而显示出成骨活性。
与不存在聚p下由acc快速形成的方解石相比,acc/聚p颗粒是生物可降解的,并且显示出优异的形态发生活性。
发明人证明,使用具有10%[w/w]聚p(“ccp10”)的acc制品,ca2+和co32-的同时释放在孵育的最初48小时期间是快速的,从而允许从支架释放生物学活性阴离子co32-和po43-。如本发明人之前所证明的那样,正磷酸盐将从聚p中酶促地释放出来(müllerweg,wangxh,diehl-seifertb,kropfk,schloβmacheru,lieberwirthi,glasserg,wiensm,
本发明方法的另一方面是,应用该方法制备具有生物活性的植入材料。本发明方法的另一方面是,应用该方法来制备刺激成骨细胞活性的人造骨植入物。此外,本文描述的本发明的另一方面是,通过应用本发明方法来制备的植入物。
用聚p稳定亚稳态acc的本发明方法,还允许将acc/聚p颗粒应用于膳食补充剂。如发明人所证明的,例如,参见图18,这些颗粒,例如,与结晶方解石多晶型物相比,“cc10”在长时间的孵育期内释放钙。
因此,本发明的acc/聚p颗粒也可以用作治疗钙缺乏症的膳食补充剂。
因此,本发明的另一方面是,稳定的acc(acc/聚p)作为膳食补充剂用于预防/治疗骨质疏松症的用途。
钙在许多生物过程(例如细胞内信号转导、肌肉收缩、神经元传递和血管收缩/血管扩张)中起重要作用。通过聚p稳定的acc可以作为钙的易于获得的食物补充剂,用于预防/治疗与钙代谢紊乱相关的许多病理学病症。
根据最近关于生物种子caco3性质的发现,可以在骨形成过程中描述通过po43-进行co32-的阴离子交换和从聚p供给正磷酸盐,接下来一系列的机制不同的过程(图4)。在骨矿物质沉积的第一阶段,像在软骨内骨化中,使包含长骨的骨骺和骨干之间的生长中心的干骺端中的软骨钙化。很可能是由ca-ii和/或ca-ix来酶促驱动这种钙化过程。第二,除了在骨形成区域和骨折部位的成骨细胞之外,积聚的血小板将聚p释放到细胞外空间,其中聚合物在正磷酸盐释放下经历alp介导的胞外水解。第三,在生物种子合成的空间附近形成的可用的磷酸盐单元,作为形成acp的来源。如本方案中所描绘的,本发明的材料是有希望的生物相容性成骨支架,其提供用于生物种子开发(caco3[co32-])和随后转化为磷酸钙(聚p[po43-])的底物。
因此,总之,本发明涉及制备具有生物活性的钛合金包被层的方法,其包括以下步骤:a)通过将聚磷酸钠(na-聚p)的水溶液与二水氯化钙(cacl2·2h2o)水溶液在形成胶体悬浮液的条件下,在升高的温度,优选在90℃下混合几小时,优选3小时来制备聚磷酸钙(ca-聚p)微粒;b)使用硅烷偶联剂将所述ca-聚p微粒胶体悬浮液偶联到合适的钛合金支架上;和c)将b)的悬浮液的ph值调节至微碱性值,优选为8.0,以允许通过ca2+离子键形成将聚磷酸盐(聚p)结合到硅烷官能化的金属支架。钛合金可以是ti-6a1-4v。硅烷偶联剂可以是(3-氨丙基)三甲氧基硅烷[aptms]。
本发明还涉及用于制备具有生物活性的非晶形聚磷酸盐取代的磷酸钙颗粒(“acap-聚p”)的方法,其包括以下步骤:a)将聚磷酸盐的水溶液加入到磷酸盐源的水溶液中;b)将所得溶液加入到溶解的钙盐中;c)将ph调节至碱性值,优选10;和d)收集、洗涤和干燥数小时后,优选在室温下24小时后形成的所得沉淀。聚磷酸盐可以是聚磷酸钠。
本发明还涉及用于制备具有生物活性的非晶形碳酸钙(acc)-聚磷酸盐微粒的方法,其包括以下步骤:a)制备在约0.1m氢氧化钠中的聚磷酸盐水溶液;b)向所述溶液中加入约0.5mol/l的碳酸钠;c)用约1.5倍体积的去离子水稀释所得溶液;d)将所述溶液与相同体积的含有氯化钙的水溶液混合,从而产生钙离子和碳酸根离子之间的约等摩尔浓度比;e)在约室温下用诸如丙酮的低级烷基酮洗涤;和f)过滤和干燥形成的沉淀物,基于磷酸盐,步骤a)中聚磷酸盐的浓度在约0.001mol/l至约1.0mol/l的范围,优选在约0.01mol/l至约0.1mol/l的范围。优选地,基于磷酸盐,步骤a)中聚磷酸盐的浓度为约0.025mol/l或约0.05mol/l。
优选的方法是,其中聚磷酸盐的链长在约3个至约1000个磷酸盐单元的范围,优选在约10个至约100个磷酸盐单元的范围,以及最优选约40个磷酸盐单元。优选的方法是,其中聚磷酸盐的量高于磷酸钙制品的5wt.%,优选10wt.%。
优选的方法是,其中钙盐是氯化钙(cacl2),磷酸盐源是磷酸氢二铵[(nh4)2hpo4]]。
在本发明中,聚磷酸钙微粒的特征在于:磷酸盐比钙的化学计量比为0.1比1至50比1,优选1比1至10比1,或化学计量比为7比1。
优选的方法是,其中计算所述钙盐的量和用作磷酸盐源的试剂的量,以获得ca/p摩尔比为10:6的磷酸钙。优选的方法是,其中聚磷酸钙微粒的平均尺寸在约0.1μm至约30μm,优选0.8μm至3μm的范围。优选的方法是,其中聚磷酸盐取代的磷酸钙颗粒(“acap-聚p”)的平均尺寸为约20nm至约300nm,优选约70nm至约120nm的范围。
优选的方法是,其还包括制备具有生物活性的钛合金植入物的步骤。还优选的方法是,其还包括制备具有生物活性的植入材料的步骤。还优选的方法是,其还包括将至少一种镓盐包含在所述植入物中的步骤。优选地,所述具有生物活性的植入材料是人造骨植入物。优选地,所述具有生物活性的植入材料是人造骨植入物。
本发明还涉及通过本发明的方法制备的植入物,或通过本发明的方法制备的稳定的acc组合物。
优选地,根据本发明制备的包被层可以用作植入物(任选地与至少一种镓盐组合使用),或作为食物或膳食补充剂(例如acc组合物)。稳定的acc组合物用于治疗钙缺乏症,或用于预防和/或治疗骨质疏松症。
现在将在以下优选实施例中进一步描述本发明,但并不受限于此。为了本发明的目的,本文引用的所有参考文献通过引用整体并入。
附图说明
图1显示从前体ca2+、po43-和oh-形成非晶形cap(acap)的示意图。acap经历了成熟为结晶ha,或者在<10wt.%的聚p存在下同样成熟为结晶cap(参见底部的插图,显示cap晶体;sem图像)。如果cap沉淀物中聚p的含量增加至>10wt.%,则形成球状非晶形acap-聚p(参见顶部的插图;sem图像)。
图2显示使用硅烷偶联剂aptms将聚p结合到钛圆片的示意图。用hc1蚀刻钛合金ti-6a1-4v,暴露于钛圆片上的羟基与硅烷偶联剂aptms交联,所述偶联剂形成至聚p的ca2+桥。脱水/缩聚后的偶联剂仍然含有游离的反应性胺基,其可用于进一步偶联至活性组分,例如,通过1-乙基-3-(3-二甲基氨丙基)碳二亚胺。在此过程中,金属表面与硅烷共价连接,而硅烷又可以通过ca2+离子桥结合聚p。
图3显示制备方解石和补充有聚p的caco3的示意图。插图显示各自产物的sem图像。
图4显示软骨内骨化过程和推荐的骨矿物质沉积阶段的示意图。穿透血管后,在骨干中的初级骨化中心的透明软骨开始钙化。在骨骺的次级骨化中心,松质骨的形成稍后开始。在骨骺和骨干之间的骨骺和骺板的表面(生长区),透明软骨的两个区域保留关节软骨。生长区的矿物沉积被细分为i期:由膜相关ca-ix介导形成的非晶形碳酸钙(acc)生物种子;ii期:在形成用于碳酸盐-磷酸盐转移反应的正磷酸盐情况下,从血小板释放的聚p经历alp介导的水解作用;和iii期:磷酸盐单元用于(碳酸化)磷酸钙形成。
图5显示与ti-ca-聚p圆片(b、d、f)相比,钛合金圆片(a、c、e)的表面粗糙度。通过光学显微镜可视化圆片的表面,并使用vk分析仪软件分析粗糙度。显示了线扫描的轨迹(c、d)。在e、f中显示代表性区域的高度剖面图;数字表示法向量直线内的偏差的最大尺寸。
图6显示通过edx光谱(a、c、e)和sem(b、d、f),对钛和ti-ca-聚p圆片进行的元素组成分析。(a、b)未处理的圆片(ti6a14v);(c、d)在聚p和cacl2反应试验中,用较低浓度的aptms(1mg/试验;聚p@ti6a14v-1)制备的ti-ca-聚p圆片;和(e,f)ti-ca-聚p圆片,其在较高aptms浓度(2mg/试验;聚p@ti6a14v-h)存在下被包被。
图7显示钛圆片对saos-2细胞生长的影响。将细胞在其他相同条件下接种到不含有钛圆片(空白柱)、钛合金圆片(交叉条纹柱)或ti-ca-聚p圆片(填充柱))的24孔板中。在1d、2d和3d的孵育期后收获细胞,并通过xtt试验确定细胞的存活力。数据表示10次独立实验的平均值±sd(*p<0.01)。
图8显示编码(a)caix和(b)alp的基因的表达。将表达值标准化为gapdh的表达。在没有任何钛圆片(空白柱),或者在钛合金圆片上(交叉条纹柱)或在ti-ca-聚p圆片上(填充柱)培养细胞。首先在mac不存在下孵育培养物3d;然后将它们转移到补充有mac的培养基中,并且如所述,继续孵育另外3d或5d。然后收集细胞,提取rna并进行qrt-pcr,以测定caix和alp转录物;将gapdh的表达作为参考。数据表示为五次独立实验的平均值±sd;每项实验一式两份进行(*p<0.01)。nd表示未检出。
图9显示具有形态发生活性的ca-聚p的钛圆片的包被层。如果包被了具有形态发生活性的ca-聚p聚合物,则金属材料(ti-6a1-4v)获得生物功能特性。在该过程中,钛表面被蚀刻,导致羟基的暴露。在ph8时,它们与硅烷偶联剂(例如aptms)形成共价连接。在这种环境下,在硅烷和聚p之间形成了ca2+离子桥。那些包被的钛圆片允许骨样saos-2细胞沉降并诱导它们进行基因表达(caix和alp);这些酶至关重要地参与骨矿物质/羟基磷灰石(ha)沉积。
图10显示从纯na-聚p“聚p”和纯“ha”以及在不同量的na-聚p存在下制备的ha(即“ha(2.5)聚p”中为2.5wt.%,“ha(5)聚p”中为5wt.%,和“ha(10)聚p”为10wt.%)获取的衍射图案。从底部到顶部给出各自的图案。“聚p”和“acap(10)聚p”没有看到衍射信号。突出显示ha或结晶cap特征的衍射峰(■)。
图11显示“聚p”样品和“ha”样品以及cap样品(其中正磷酸盐已被聚p部分取代,“ha(2.5)聚p”样品、“ha(5)聚p”和“acap(10)聚p”样品)的ftir光谱。标记了“co32-和po43-”的一些振动带;另外指出了h2o和co2带的区域。(a)在4000至500的波数范围(cm-1);(b)2000cm-1至500cm-1部分的放大。
图12显示聚p和cap颗粒的tem显微照片。(a)“ha”晶体;(b和c)“ha(2.5)聚p”晶体和“ha(5)聚p”晶体;和(d)“acap(10)聚p”非晶形球状颗粒。
图13显示在saos-2细胞中编码(a)ⅰ型胶原蛋白(col-i)或(b)碱性磷酸酶(alp)的基因的稳态表达水平。将细胞暴露于10μg/lml的聚p纳米颗粒“aca-聚p-np”(填充柱),或暴露于100mg/ml的“ha”(空白柱),“ha(2.5)聚p”(右阴影柱),“ha(5)聚p”(左阴影线柱)或“acap(10)聚p”(交叉阴影线柱)。在标准培养基/血清中初始孵育3d后,将细胞转移到缺乏(减去mac)或含mac(加上mac)的培养基/血清中。在7d的孵育期后,收获细胞,提取rna,随后用于qrt-pcr分析。表达值以与参考基因gapdh的比值给出。结果是5次平行实验的平均值;*p<0.01。
图14显示方解石以及“ccp5”(0.05gna-聚p/试验)和“ccp10”(0.1gna-聚p))的ftir光谱。将方解石与acc的主要区别振动区域/信号,o-h(约3250cm-1)的振动范围和在725cm-1处的co3的不对称v2线加画圆圈。
图15显示从(a)方解石和(b)含有两种不同浓度的聚p的两种caco3制品(“ccp5”或“ccp10”)获得的xrd图案。特征信号被突出显示并用各自的米勒指数标记,在括号中给出。请注意在(a)和(b)之间的纵坐标标注的不同比例。
图16显示由cacl2·2h2o和na2co3形成的固体的形态;sem分析。(a和b)在不存在聚p的情况下,形成方解石晶体。在沉淀过程中加入聚p后,形态发生变化。(c和d)在存在5%聚p的情况下,“ccp5”颗粒显示出球形外观。(e和f)在10%聚p的情况下,“ccp10”,固体显示出对应于球霰石晶体(vat)的薄片状形状。
图17显示在3d孵育期后,在50μg/ml的“ccp10”(a和b)或方解石(c和d)存在下,saos-2细胞的生长方式。通过相位差/nomarski光学鉴定细胞。试验中的caco3颗粒在相差图像中变得可见,并被标记(><)。
图18显示从caco3颗粒释放ca2+。将“ccp10”或方解石在tris-hcl缓冲液(ph7.4)中孵育不同时间,分析上清液中ca2+浓度。结果是6次平行实验的平均值;*p<0.01。
图19显示在(a)saos-2细胞和(b)msc中alp基因的稳态表达水平。细胞保持在没有任何caco3固体的情况下(对照),或暴露于50μg/ml的“ccp5”(左侧阴影线柱)、“ccp10”(右侧阴影线柱)或方解石(填充柱)。在mac不存在下的3d预孵育期之后,在mac不存在(减去mac)或暴露于mac(加上mac)的情况下,继续孵育细胞。在7d孵育后收获细胞,提取其rna并进行qrt-pcr分析。表达值以与参考基因gapdh的比值给出。结果是5次平行实验的平均值;*p<0.01;根据加晶种期间在细胞中测量的表达来计算所述值。
图20显示在“ccp10”和聚p(ca2+复合物)存在下,在saos-2细胞中bmp2基因的稳态表达水平。细胞保持在没有任何添加剂的情况下(对照),或暴露于50μg/ml的“ccp10”(右阴影线柱)、5μg/ml的聚p(ca2+络合物;50μm磷酸盐单元;交叉阴影线柱)或50μg/ml方解石(填充柱)。在mac不存在下的3d预孵育期之后,在mac存在下将细胞继续孵育长达7天,并通过qrt-pcr分析bmp2表达。表达值以与参考基因gapdh的比值给出。结果是5次平行实验的平均值;*p<0.01;根据加晶种期间细胞中测定的表达来计算所述值(第0天);#p<0.01(仅适用于“ccp10”);在相应的孵育期间,根据在具有聚p(ca2+络合物)的细胞中测量的表达来计算所述值。
图21显示微球的形态;(a)对照球“cont-mic”,和(b)加载聚p的球体“聚p-mic”。
实施例
在以下实施例中,仅针对链长为40个磷酸盐单元的聚p分子描述本发明的方法。通过使用具有较低和较高链长(如约20个至100个单元)的聚p分子可获得类似的结果。
钛-ca-聚p(ti-ca-聚p)圆片
钛合金(ti-6a1-4v)圆片被蚀刻,以允许与硅烷偶联剂aptms交联(图2)。在第二步,通过ca2+离子桥用聚p覆盖圆片。最后将样本ti-ca-聚p圆片在100℃下干燥。我们目的是使用硅烷偶联剂aptms来提供另外的官能团(胺基团),以将生物活性肽偶联至聚p包被的金属表面。钛圆片的功能化也用3-(三甲氧基硅基)丙基甲基丙烯酸酯成功地进行,仅允许聚p-钛包被层。
钛合金圆片与ti-ca-聚p圆片之间的比较(光学显微镜图像)显示,与钛合金圆片的深灰色表面颜色相反,ti-ca-聚p圆片具有闪亮的银白色外观。在用聚p包被圆片的表面之后,它们失去其高的粗糙度。虽然未处理的圆片具有≈5.5μm的平均粗糙度,最大值为7.02μm(图5a、图5c、图5e),但是聚p包被的圆片最大程度地暴露了0.78μm的表面粗糙度(图5b、图5d、图5f)。
通过edx光谱进行钛圆片表面的元素特异性分析(图6)。未经处理的圆片的表面,在4.5kev显示出钛的优势kα峰,并在4.9kev显示出较低kβ峰(图6a)。表面的形态被标记为粗糙(图6b)。如果在有聚p和cacl2的包被溶液中孵育后,蚀刻并与较低浓度的aptms(1mg/试验)反应后检查钛圆片,通过sem可解析ca-聚p微粒(müllerweg,tolbae,
ca-聚p包被层的耐久性
如下文“方法”部分所述,通过测定来自sbf(缺少ca2+作为组分)中包被圆片的ca2+释放,来测量聚p的表面包被层。在平行试验中,测量了来自ti-ca-聚p圆片以及未处理的钛圆片(对照)的ca2+释放。作为另外的对照,将一ti-ca-聚p圆片插入补充有1μg/ml的alp的sbf培养基中;将所有样品于37℃孵育。在所有三种试验中的时间为零时,ca2+浓度为<3μg/ml。在孵育培养基中1d后,测定的ca2+量如下:ti-ca-聚p圆片:<3μg/ml(<3μg/ml[对照];12±3μg/ml[ti-ca-聚p圆片+alp]);进行5次平行试验。与和alp一起的ti-ca-聚p圆片的试验相反,在3d的孵育期后,在含有ti-ca-聚p圆片的试验中ca2+释放略微增加。测量以下值:5.2±0.8μg/ml[ti-ca-聚p圆片](<3μg/ml[对照];86.9±3.2μg/ml[ti-ca-聚p圆片+alp])。在孵育试验12d后,其值如下:9.7±1.2μg/ml[ti-ca-聚p圆片];<3μg/ml[对照];153.1±17.1μg/ml[ti-ca-聚p圆片+alp]。
saos-2细胞在钛圆片上的生长
通过如下文“方法”部分中所述的xtt试验,来测定骨样saos-2细胞的总体生长速率。对于所有三个平行的实验系列,将细胞以3×104个细胞/孔(2ml试验)的密度接种;不含钛圆片的试验、钛合金圆片试验、ti-ca-聚p圆片试验(图7)。在1-d孵育期后,细胞密度从0.3吸光度单位增加到0.49±0.6单位(无圆片试验)和0.47±0.05单位(含ti-ca-聚p圆片),而在有钛合金圆片的试验中密度下降至0.26±0.03单位。该趋势在随后的孵育期内增加,并且在3-d孵育期后所述值达到:0.09±0.02(钛合金圆片;显著降低)、0.72±0.08(无圆片)和0.68±0.07(ti-ca-聚p圆片)。这些数据意味着钛表面不支持saos-2细胞的生长,而在ti-ca-聚p圆片上生长的细胞显示与没有任何圆片的培养物相同的生长动力学。
通过染色圆片表面,还强调了包被有聚p的圆片的特性,其是用于saos-2细胞生长的非常合适的基底。未包被聚p的钛合金圆片与saos-2细胞孵育3d;在此之后,没有在圆片上看到细胞。相比之下,如果将聚p包被的ti-ca-聚p圆片在saos-2细胞存在下孵育相同的时间段,则观察到几乎均匀的单细胞层。在较高放大率下仔细检查显示,细胞显示出细胞扩散的特性,这是细胞至关重要的生存和生长的特征信号。
作为对saos-2细胞容易生长到ti-ca-聚p圆片上的结论的进一步支持,分析了覆盖有细胞区域的元素碳(c)、钛(ti)和磷(p)的分布。通过基于sem的edx映射,进行元素的半定量测定。通过记录背散射电子获得细胞的定位。在细胞生长区域内,测量元素c的高积聚,在细胞生长的圆片的周围表面上,细胞区域外部的钛和聚p显著。
碳酸酐酶ix和碱性磷酸酶的表达
作为生长在钛圆片上的saos-2细胞功能活性的标记物,通过定量qrt-pcr测定了编码酶碳酸酐酶ix(caix)和碱性磷酸酶(alp)的两个基因的表达。在mac不存在下生长3d的saos-2细胞中caix转录物的稳态水平的研究,对于含有钛合金圆片的培养物,表达水平从0.31±0.03(在接种时间)到0.12±0.01显著降低,而不存在圆片或存在ti-ca-聚p圆片的培养细胞中的水平分别从0.27±0.02和0.25±0.03增加到0.38±0.04和0.40±0.05(图8a)。随后在mac存在下孵育培养物,导致在不含圆片或ti-ca-聚p圆片浸没的试验中caix表达水平的增加。在mac存在下5d后,检测到在不存在圆片的细胞中,caix转录物水平从0.38±0.04至0.81±0.08显著增加。在ti-ca-聚p圆片上培养的细胞中也发现该基因表达从0.41±0.05至0.59±0.06显著增加。这些结果强调,用聚p包被到钛植入材料上提供了具有生物活性表面的材料(图9)。
同时,同样通过qrt-pcr来测定酶alp基因的表达。再次,数据(图8b)显示,在3d孵育期后,相对于不存在任何圆片,在含有钛合金圆片的培养物中alp的表达水平显著降低。稍后在孵育过程中,水平低至表达无法可靠地记录。相比之下,在mac存在下alp的稳态表达,在无圆片的试验中和在有ti-ca-聚p圆片的试验中都显著增加,分别为从0.038±0.005到0.097±0.007(在没有圆片的第5天),从0.034±0.004到0.074±0.007。
在另一组实验中,在100μm硝酸镓存在下进行测定(参见表1)。如上所述,在没有任何钛圆片、或在钛合金圆片上、或在ti-ca-聚p圆片上培养细胞,首先在mac不存在下培养3d,然后在补充有mac的培养基中再培养5d。结果表明,与硝酸镓不存在下为0.27±0.02至0.81±0.08(另见图8)相比,在圆片不存在下,在mac不存在下生长3d,以及随后在mac存在下生长5d的saos-2细胞中,caix转录物的稳态水平从0.24±0.05增加至0.89±0.11(表1)。如果将细胞在ti-ca-聚p圆片上培养,发现与不存在硝酸镓的试验相比,在硝酸镓存在下caix转录物的水平更强的增加;相对于从0.25±0.04至0.59±0.06(不存在硝酸镓;还参见图8),caix转录物的稳态水平从0.27±0.04增加到0.96±0.15(存在100μm硝酸镓),表明ca-聚p包被层和镓盐有很强的协同效应(与没有圆片和有ti-ca-聚p圆片的试验比较)。
如果在不存在圆片和存在钛合金圆片或ti-ca-聚p圆片的情况下,测定了硝酸镓对alp转录物稳态水平的影响,则得到类似的结果。在钛圆片不存在下添加镓盐,与没有这一添加剂的试验相比,对alp转录物水平的影响很小;而在ti-ca-聚p圆片存在下,如果与没有该补充物的试验(从0.030±0.003增加到0.074±0.007;还见图8)相比,也观察到对ca-聚p引起的alp转录物水平增加的强协同效应(从0.027±0.003增加到0.115±0.007)。在未包被的钛合金圆片上培养的细胞,没有观察到可检测的或仅观察到非常小的转录物水平。
表1.镓对编码caix和alp的基因表达的影响。在试验混合物中不存在或存在100μm硝酸镓时,如图8的图例所述进行实验。将表达值标准化为gapdh的表达。在没有任何钛圆片、或者在钛合金圆片、或者在ti-ca-聚p圆片上培养细胞。首先将培养物在mac不存在下孵育3d,然后转移到补充有mac的培养基中,继续孵育另外3d或5d。nd,不可检测。
xrd分析
通过应用粉末x射线衍射(xrd)法,进行“ha”以及聚p-ha颗粒的相鉴定(图10)。而对于纯na-聚p而言,没有明确的衍射信号可以被解析,表明是非晶形相,纯的“ha”以及“ha(2.5)聚p”和“ha(5)聚p”表现出宽的衍射峰,表明形成低结晶度的ha;没有检测到其他结晶相(jcpds[http://www.icdd.com/]#09-0432)。然而,当聚p的量增加至10wt.%时,如在“acap(10)聚p”中,在xrd图案中没有看到结晶的迹象(图10)。这些结果表明,随着聚p含量的增加,制备的ha样品的结晶度逐渐降低。
fitr光谱分析
在这里记录的cap的所有光谱[如纯“ha”以及“ha(2.5)聚p”和“ha(5)聚p”],显示了典型的ha条带(图11a和图11b),除了“acap(10)聚p”。如预期的那样,在1090cm-1,1015cm-1和960cm-1处记录了具有高强度的ha的典型吸收带,具有对称v1(po43-)伸缩振动和不对称v3(po43-)伸缩振动。此外,在556cm-1和604cm-1处的v4(po43-)峰是特征弯曲振动。相比之下,“acap(10)聚p”的光谱显示了在1100-900cm-1的区域中对称v1(po43-)伸缩振动和不对称v3(po43-)伸缩振动以及出现为以610cm-1附近为中心的一个峰的p=o弯曲振动的v4(po43-)谐波的磷酸盐吸收带的明显移动。另外,从~3600cm-1直至3100cm-1的范围的宽吸收带指向v3和v1,具有拉伸模式的与氢键合的h2o分子。在1629cm-1处的吸收带归因于h2o分子的变形模式v2,证明在合成样品中物理吸附水的存在。据报道,cap的ftir光谱中556cm-1和604cm-1附近的振动带,反映了结晶po43-的谐波振动的特征弯曲信号;两个峰的移位表明从结晶相向非晶形相的转变。在“acap(10)聚p”的图中清楚地看出这种移动,其中两个峰现在显示为一个峰,表明该样品的非晶形性质。这个发现也与报道的xrd图案一致(图10)。
为了比较,聚p的谱也包括在cap追踪中(图11)。显然的,对于聚p,1261cm-1附近的峰显示属于体现聚p特征的(o-p=o)的不对称拉伸模式。接近1090cm-1和960cm-1的吸收带分别属于(o-p-o)的不对称拉伸模式和对称拉伸模式。这些信号进一步证实了聚p的存在。此外,864cm-1附近的吸收带指示了p-o-p连接的不对称拉伸模式,和以763cm-1附近为中心的部分分裂带应属于这些连接的对称拉伸模式。
tem和粒度分布结果
通过tem分析cap样品的形态。“ha”样品显示平均长度为39±8nm和宽度为14±4nm的针状纳米晶体(图12a)。在长度为42±10nm和宽度为9±5nm的“ha(2.5)聚p”样品中,可以看出几乎相同的尺寸(图12b)。“ha(5)聚p”制品中存在的晶体稍长为56±12nm且宽度为6±3nm(图12c)。相比之下,含有最高比例聚p的cap制品(“acap(10)聚p”)显示出具有不同形态的颗粒(图12d)。不同于解析的直径为70nm至120nm(96±15nm)的针状结构球形颗粒。那些颗粒具有聚集成较大实体的倾向。
cap样品和ca-聚p纳米颗粒对细胞生长的影响
通过应用mtt试验来检测在cap样品上的saos-2细胞的细胞存活力和生长。将那些样品以100μg/ml的浓度加入细胞。并行地,用10μg/ml的ca-聚p纳米颗粒“aca-聚p-np”进行孵育,所述“aca-聚p-np”是经证明可增加细胞生长速率并导致alp和col-i基因表达增加的样品。
结果表明,在2d的孵育期后,在不同基底上没有看到细胞生长的显著差异。然而,在3d的孵育期后,在“aca-聚p-np”存在下测量到saos-2细胞生长的显著增加(从1.74±0.19吸光度单位[第2天]至2.45±0.20吸光度单位)。在不同cap样品上生长的增加较低,对于“ha”(从1.54±0.18至1.93±0.21)和“acap(10)聚p”(从1.54±0.18至2.31±0.25)生长的增加是显著的。
sem分析
将细胞在纯“ha”上或在“acap(10)聚p”圆片上培养3d。然后将样品用多聚甲醛固定,并通过sem检查。可以看出,在两种试验中,对于“ha”和“acap(10)聚p”培养物,细胞均牢固地附着于基底。在较高放大倍数下,细胞扩散的特性变得明显。
cap上的saos-2细胞的基因表达倾向
最初将骨相关saos-2细胞培养3d,然后转移到新的缺少mac或补充mac并且还含有cap样品(100μg/ml)或聚p纳米颗粒(10μg/ml)培养基中。然后在qrt-pcr分析之前继续孵育7d,以确定col-1或alp转录物的稳态水平(图13)。
测定显示,在接种细胞时col-1的表达低且为0.26±0.07个表达单位,其与gapdh的表达相关。在不存在mac和存在cap样品或聚p纳米颗粒7d的情况下,与“ha(2.5)聚p”(至0.35±0.05)、“ha(5)聚p”(至0.43±0.05)以及“acap(10)聚p”(至0.52±0.06)孵育之后,表达水平显著增加,如预期的那样,对于聚p“aca-聚p-np”(至0.63±0.07)也是一样;图13a。暴露于mac后,所有稳态表达水平显著较高,并且例如“ha”的值达到0.41±0.05,和“acap(10)聚p”的值达到1.06±0.11。后者“acap(10)聚p”的值接近暴露于聚p纳米颗粒的细胞中具有的1.38±0.16的诱导水平。
如果与参考基因gapdh相关,则记录alp基因相当的诱导表达模式。再次,与在mac存在下孵育7d的细胞的测量值相比,在mac不存在下alp表达水平较低(图13b)。只有在没有mac但补充有聚p纳米颗粒的试验中,alp的表达水平的增加是显著的(从0.12±0.02到0.17±0.01)。然而,暴露于mac后,所有含聚p的cap制品的表达水平显著高于细胞接种期间所见到的表达水平。对于“ha(2.5)聚p”的增加值为0.15±0.03,对于“ha(5)聚p”的增加值为0.28±0.03,和对于“acap(10)聚p”的增加值为0.89±0.09。再次,如果与含有较少量聚p的样品相比,后一种表达水平更接近于具有1.37±0.16的暴露于聚p的细胞所测定的值。
聚p对方解石形成的影响:ftir光谱和xrd光谱
对于所有的caco3固体,记录以下ftir信号:在≈1080cm-1处的v1(对称伸缩),在≈870cm-1处的v2(平面外弯曲),在≈1400cm-1处的v3(双重退化平面不对称伸缩)和在≈700cm-1处的v4(双重退化平面弯曲)。用ftir/kbr小球获得的发表的ir数据(rodriguez-biancojd,shaws和benninglg(2011)thekineticsandmechanismsofamorphouscalciumcarbonate(acc)crystallizationtocalcite,viavaterite.nanoscale3:265-271),包括方解石的位于约1400cm-1(v3)的峰、876cm-1(v2)的峰和714cm-1(v4)的峰,以及球霰石的位于1090cm-1(v1)的峰、870cm-1(v2)的峰和745cm-1(v4)的峰(图14)。在聚p不存在下制备的我们的样品的特征如下。对于方解石,记录了典型的振动带1391cm-1、872cm-1和712cm-1,而在聚p存在下制备的样品,对于“ccp5”聚p在1398cm-1、869cm-1和742cm-1处显示出吸附峰,以及对于“ccp10”显示在1398cm-1、869cm-1和741cm-1处的带,证明了球霰石的形成。显然,在制备的caco3固体中(“ccp10”对“ccp5”),聚p含量较高时,对于球霰石在741cm-1附近的信号强度降低。这表明acc的形成。除co32-的吸收峰外,从1200cm-1到950cm-1的峰对应于聚p中磷酸盐的吸收峰。
用xrd证实了上述结果,其中给出了在聚p不存在下制备的样品在大约23°、30°、36°和40°处的衍射峰;那些信号对应于方解石。相反,在聚p(“ccp5”)存在下制备的样品在大约24°、27°、32°和44°处显示出峰,这也反映了球霰石的存在。此外,这些数据证明,在聚p不存在下制备的caco3固体是纯方解石(图15a),而“ccp5”样品由与acc结合的球霰石组成,其可以从样品“ccp5”的低强度信号以及衍射峰的加宽来推理(图15b)。因此,如“ccp10”中聚p的量的增加,降低了acc向球霰石的转化率。明显地,来自“ccp10”样品的xrd图案,其显示了样品的非晶形性质,但是还含有少量的球霰石。
形成的固体形态
通过sem研究了由cacl2·2h2o和na2co3沉淀形成的固体。在聚p不存在下形成的颗粒其显微照片显示典型的结晶方解石,由{104}面包围的菱面体晶体,图16a和图16b。颗粒的尺寸在5.3μm至8.9±2.4μm之间变化。相反,在聚p存在下由cacl2·2h2o和na2co3形成的固体显示不同的形态。在较低的聚p浓度下,“ccp5”颗粒呈球形外观,球形晶体的平均尺寸为9.4±3.7μm(图16c和图16d);我们将这些颗粒归属于球霰石。它们被非常丰富地积累的尺寸范围为100nm至200nm的小纳米颗粒包围,我们将其指定为acc。如“ccp10”中增加聚p,球状颗粒消失,并被五角形片状/六边形片状的颗粒(5-10μm大小的球霰石)取代(图16e和16f)。
caco3样品对细胞生长/存活力的影响
通过应用mtt试验(参见上文)测定暴露于caco3制品后saos-2细胞的细胞生长/存活力。将caco3样品以50μg/ml的浓度加入到细胞中。并行地,进行缺乏任何caco3固体的对照试验。结果表明,在对照试验和三种caco3系列(“ccp5”、“ccp10”或方解石)中,细胞生长/存活力从时间0的0.70±0.11增加到2d后的大约1.1个吸收单位和3d后的2.35个单位,仅有不显著的变化。
caco3固体在培养基中的稳定性
saos-2细胞以附着方式生长(pautkec,etal.(2004)characterizationofosteosarcomacelllinesmg-63,saos-2andu-2osincomparisontohumanosteoblasts.anticancerres24:3743-3748)。如果培养物暴露于方解石或“ccp5”固体,培养皿表面上的生长行为,在含有“ccp10”(图17a和图17b)或方解石(图17c和图17d)的试验中是类似的。3d后,细胞生长几乎达到融合。然而,值得注意的是,与在方解石试验中观察到的那些相比,在含有“ccp10”的试验中,在这段时间后漂浮在培养基中的矿物颗粒的数量显著降低。该观察结果可作为“ccp10”颗粒在5d孵育期内经历溶解的指示。这一发现由测定支持,其显示在模拟体液(oyanea,kimhm,furuyat,kokubot,miyazakit,nakamurat(2003)preparationandassessmentofrevisedsimulatedbodyfluids.jbiomedmaterresa65:188-195)中的3d孵育期后,基于可沉淀的碳酸盐测量(数据未显示),方解石颗粒的量仅降低5-10%,而只有35%的“ccp10”颗粒可被回收。
从caco3颗粒释放ca2+
在单独的试验中,将方解石或“ccp10”加入用1mtris-hcl(ph7.4)缓冲的1ml试验中。虽然几乎没有从方解石样品释放ca2+,但在48hr后,已经从“ccp10”释放了6.8±1.1μg/ml(反应混合物中总ca2+的68%);在整个192hr孵化期间,这一程度进一步增加(图18)。
在saos-2细胞以及msc中alp的表达
在不存在和存在mac的情况下,测定caco3样品对saos-2细胞以及msc的形态发生活性。使用saos-2细胞,确定在mac不存在下,alp与参考基因表达(gapdh)之间的表达比显著地从0.31±0.9增加到≈0.6。在没有mac的实验组中,没有测量到显著差异,而不管在试验中caco3样品是不存在(对照)还是存在(图19a)。然而,如果在mac激活的细胞中测定表达比(alp:gapdh),则测量到比值显著增加至0.87±0.12(在对照中)、增加至1.74±0.23(“ccp5”)、或增加至1.86±0.29(“ccp10”)。相反,在用方解石的试验中,没有测量到细胞的响应(0.14±0.05)。
如果将msc用于实验,则发现如果与参考gapdh基因表达相关的alp的类似表达模式。再次,在任何caco3固体不存在下以及在“ccp5”和“ccp10”两者存在下的试验中,在mac存在下看到显著增加的表达比。在暴露于方解石的细胞中没有测定到诱导作用(图19b)
在saos-2细胞中bmp2的表达
通过qrt-pcr分析测定bmp2对“ccp10”和聚p(ca2+络合物)响应的表达水平。将saos-2细胞在矿化培养基(mccoy's培养基/mac)中孵育长达7天。在实验开始时,向培养物中加入“ccp10”(50μg/ml)、聚p(ca2+络合物;5μg/ml;对应于磷酸盐的50μm)或方解石(50μg/ml)。终止后,从培养物中提取rna并进行qrt-pcr。使用管家基因gapdh的表达作为参考。如图20所示,加入“ccp10”或聚p(ca2+络合物)3至7天后,bmp2的表达水平显着增加。然而,与聚p(ca2+络合物)相比,“ccp10”的bmbp2表达增加更快。在“ccp10”的3天孵育期后,已经达到bmb2基因表达的最高水平,而由聚p(ca2+络合物)诱导的该基因的表达仅在更长时间(5天孵育期)后达到最高水平(与“ccp10”相比类似的水平)。在第3天,与聚p(ca2+络合物)相比,响应“ccp10”的bmp2的表达水平显著高(约2倍),表明两种组分的“协同”效应。7天后,“ccp10”和聚p(ca2+络合物)的表达水平均降低,但依然保持显著。在聚p不存在下,由亚稳态acc形成的方解石对bmp2基因表达没有显示任何刺激作用。
用于动物研究的微球
对照球“contmic”具有(≈845μm[820±60μm];n=50)的大小,而含有聚p的那些大小稍微小一些(≈838μm[816±65μm]);图21a和图21b。微球表面的结构是多孔的,具有25nm-30nm的孔(此处未示出)。“聚p-mic”中的聚p含量为7.26±0.92%。测定“contmic”和“聚p-mic”的颗粒硬度;“contmic”的中值redym刚度为26.99±6.22kpa,“聚p-mic”微球的中值redym刚度为23.96±23.96kpa。
大鼠中的相容性研究
如“材料和方法”所述,将微球样品(20mg),“contmic”和“聚p-mic”嵌入大鼠背部的肌肉中。在2周、4周或8周后,取出带微球的组织样品,切片并用苏木精溶液染色。在用于“contmic”和“聚p-mic”系列的每组所有三只处死的实验室动物中,没有在一个离体的标本中可以看到任何组织病理学改变的迹象。2周后,在微球已经放入肌肉的区域,一些细胞分散在微球区域内。然而,“cont-mic”微球在肌肉区域中停留4周和8周后,它们显示是空的或接近无细胞的。相比之下,在“聚p-mic”微球内4周后,细胞在球体内的积累明显。8周后,球体几乎充满了浸润细胞。
8周后植入区域硬度的测定显示了明显增加的中值redym刚度,对于“cont-mic”是33.13±7.97kpa,对于“聚p-mic”微球是60.11±12.13kpa。在植入前,大鼠背部肌肉具有74.40±14.33kpa的中值redym刚度。
方法
聚磷酸盐
已从chemischefabrikbudenheim(budenheim;德国)获得实施例中使用的聚磷酸钠(平均链为40个磷酸盐单元的na-聚p)。
ti-ca-聚p圆片
钛合金(ti-6a1-4v)圆片(直径15mm和厚度2mm),可以从例如nobelbiocare获得。使用前,用砂纸(碳化硅,matador)抛光,然后在蒸馏水中用超声波清洗,随后在丙酮(10min)和40%乙醇溶液(15min)中洗涤,最后在蒸馏水中漂洗20min。将样品在50℃下干燥24小时。然后在室温下将钛合金圆片在20ml的5mhcl中孵育6h。在蒸馏水中温和洗涤之后,将圆片在室温下干燥,并在硅烷偶联剂(3-氨丙基)三甲氧基硅烷[aptms](例如,来自sigma-aldrich)存在下,将处理的圆片样品用10mlca-聚p纳米颗粒悬浮液覆盖。
通过将0.5gna-聚p与atpms溶液(1wt%)在100ml水中混合,然后加入0.1gca2+氯二水合物(cacl2·2h2o)来制备ca-聚p微粒。将钛圆片于90℃在上述悬浮液中孵育3小时;在这些条件下,最初形成胶体悬浮液。将环境ph调节至8.0,以便通过ca2+离子键/桥接将聚p结合到硅烷蚀刻的钛圆片。样品保留在该悬浮液中1d。研究了两种不同的atmps浓度(分别为1mg/试验和2mg/试验)对在钛表面上形成的包被层形态的影响。最后,取出样本钛-钙-聚p(ti-ca-聚p)圆片,并在100℃干燥(参见图1)。
在实施例中描述的实验中,如果没有提及其他的,则使用以较高比例的aptms然后与ca-聚p一起制备的圆片。
ha和聚p-羟基磷灰石的合成
可以通过湿法化学沉淀法,由氯化钙(cacl2)作为ca2+源和磷酸氢二铵((nh4)2hpo4)作为磷酸盐源合成羟基磷灰石(ha)纳米粒子。为了沉淀化学计量的ha(ca10(po4)6(oh)2;ca/p比为1.667),将100ml0.3m(nh4)2hpo4水溶液逐滴加到100ml0.5mcacl2水溶液中。为了获得ca/p摩尔比为10:6的ha,计算试剂的量。加入氢氧化钠溶液,使反应的ph值保持在10。
为了制备各种聚p含量的聚p取代的ha纳米颗粒,起始成分(cacl2和(nh4)2hpo4)还如下补充有2.5wt.%、5wt.%或10wt.%的na-聚p(称为ha,或各自的cap制品)。将各自量的na-聚p、0.12g[“ha(2.5)聚p”]、0.25g[“ha(5)聚p”]或0.50g[“acap(10)聚p”]加入到(nh4)2hpo4水溶液;然后将该溶液加入到溶解的cacl2中。将ph保持在10。将所得沉淀物在室温下放置24h。然后过滤沉淀,用蒸馏水洗涤3次,然后在60℃的热风烘箱中干燥24h。最终粉末称为“ha”、“ha(2.5)聚p”、“ha(5)聚p”和“acap(10)聚p”。
制备聚p纳米颗粒
对于比较功能/生物学研究,可以如所述制备非晶形ca-聚p纳米颗粒(müllerweg,tolbae,
制备碳酸钙微粒
使用等摩尔浓度比ca2+和co32-的cacl2·2h2o溶液和na2co3溶液,通过快速混合在水溶液(室温)中直接沉淀制备碳酸钙(caco3),示意图见图3。
为了研究聚p对沉淀的caco3的影响,向其中补充有0.05g或0.1gna-聚p的20ml0.1mnaoh的溶液中加入1.05gna2co3;随后将该溶液用30ml去离子水稀释。然后加入含有1.47gcacl2·2h2o的50ml水。由此,分别向caco3沉淀试验中加入5%[w/w](加入0.05gna-聚p)和10%[w/w](0.1gna-聚p)聚p。将获得的悬浮液过滤,用丙酮洗涤并在室温下干燥。样品称为“ccp5”(每次caco3沉淀试验为0.05gna-聚p)或“ccp10”(0.1g)。
ca-聚p包被层的耐久性
可以例如通过测定来自圆片的ca2+释放,来量化钛圆片周围的ca-聚p包被层的稳定性和耐久性。将对照圆片以及ti-ca-聚p盘浸没在模拟体液(sbf)中,但是省略ca2+作为组分;将ph调节至8.0。试验体积为1ml,并且在37℃下进行孵育。通过应用络合滴定法测定ca2+浓度;使用试剂eriochromeblackt(例如,来自sigma-aldrich)。在实施例中描述的实验中,在圆片一个平面上的聚p包被层的表面厚度,已通过显微镜确定为≈5μm。反过来,在圆片一个平面上的ca-聚p(密度≈3g/ml)的总量,具有≈2.4mg的值。
如实施例所示,将5μg来自牛肠粘膜的碱性磷酸酶(alp)(例如来自sigma;≥6,500dea单位/mg蛋白)加入到反应混合物中。
显微镜分析
可以例如用来自keyence的、配备有vh-z25变焦镜头(25x至175x放大)或vh-z-100长距离高性能变焦镜头(高达1000x放大)的vhx-600数码显微镜,来进行圆片的光学显微镜检查。可以例如通过使用keyencevk分析仪软件,来测量表面粗糙度。对于扫描电子显微镜(sem)分析,可以使用例如hitachisu8000电子显微镜(hitachihigh-technologieseuropegmbh,krefeld,德国)。
电子显微镜和能量色散x射线光谱
对于透射电子显微镜(tem)分析,例如可以使用在120kv的加速电压下在tecnai12透射电子显微镜(fei)上操作的temcam-f416(4k×4k)ccd照相机(tvips)。该设备与粒度分析仪(imagej)连接;在实施例中描述的实验中,已经评估了25-50个晶体/球体。
可以例如在低电压(1kv)用su8000仪器(hitachihigh-technologieseurope),进行扫描电子显微镜(sem)分析。对于实施例中所述的研究,使细胞于6孔板中在已经压制成直径为34mm、厚1mm圆片的cap制品上生长3天。用4%多聚甲醛固定在cap基底上生长的细胞。
可以例如用连接到扫描电子显微镜(nova600nanolab;fei),在10kv操作且收集时间为30-45s的edaxgenesisedx系统,进行能量色散x射线(edx)光谱。分析大约5μm2的区域。
可以用例如hitachisu8000显微镜,在低电压(<1kv,近表面有机表面的分析)下进行edx映射。将sem与xflash5010检测器(允许同时进行基于edx的元素分析的x射线检测器)相结合。设备的这种组合用于更高电压(10kv)分析,在此期间将xflash5010检测器用于沉积物表面的元素映射。hypermap数据库用于解释。
x射线衍射分析
可以如所述进行x射线衍射(xrd)实验(raynauds,etal.calciumphosphateapatiteswithvariableca/patomicratioi.synthesis,characterizationandthermalstabilityofpowder.biomaterials2002;23:1065-1072)。可以例如在具有cukα辐射(
傅里叶变换红外光谱
可以通过使用微磨(玛瑙研钵和杵)矿物粉末,例如在配备有goldengateatr部件(specac)的atr-ftir分光镜/varian660-ir光谱仪(agilent)中,进行傅立叶变换红外(ftir)光谱分析。实施例中所示的每个光谱代表具有4cm-1(通常为550-1800cm-1)的光谱分辨率的100次扫描的平均值。可以例如用varian660-ir软件包5.2.0(agilent),实现光谱的基线校正、平滑化和分析。可以例如用originpro(版本8.5.1;originlab)进行光谱的图形显示和注释。
从caco3颗粒释放ca2+
在单独的试验中,将100μg/ml方解石或“ccp10”加入到含有1ml1mtris-hcl(ph7.4)的eppendorf管中。在室温下孵育2h、2d、3d和8d后,取出100μl的样品并离心,分析上清液中ca2+浓度。可以例如用光度测试试剂盒(例如,millipore/merckchemicals;货号100858“calciumcelltest”)进行测定。从测试试验中减去空白值。
培养saos-2细胞
将骨细胞如saos-2细胞(人类成骨肉瘤细胞),在补充有2mml-谷氨酰胺、10%或15%热灭活胎牛血清(fcs)和100单位/ml青霉素和100μg/ml链霉素的mccoy培养基(biochrom-seromed)中培养。将细胞在25-cm2培养瓶或6孔板(表面积9.46cm2;例如来自orangescientifique)中,在37℃的湿润培养箱中孵育。常规地,以3×104个或1×104个细胞/孔开始培养,总体积为3ml。在此指出,培养物首先在不存在矿化-激活混合物(mac)中,在包含5mmβ-甘油磷酸酯、50mm抗坏血酸和10nm地塞米松的情况下培养3d。然后在不存在或存在mac的情况下,将培养物继续孵育多达7d。在实验开始时,向每个孔中加入ha/聚p矿物样品(100μg/ml[ha,cap]或10μg/ml[“aca-聚p-np”])。每三天,用新鲜的培养基/血清替换培养基,并且在此指出也有mac。对于使用圆片的研究,使用24孔板(例如,来自corning;每个孔的直径为15.6mm),其中插入15mm大的圆片。以2ml细胞/培养基/fcs的总体积进行试验。
在实施例中所示的另一系列实验中,在100μm硝酸镓存在下进行了试验。
细胞增殖/细胞存活力试验
可以例如通过基于四唑盐xtt(例如,细胞增殖试剂盒ii(roche))或3-[4,5-二甲基噻唑-2-基]-2,5-二苯基四唑(mtt;#m2128,sigma)的比色测定法,来测定细胞增殖/生长(wangxh,etal.(2014)modulationoftheinitialmineralizationprocessofsaos-2cellsbycarbonicanhydraseactivatorsandpolyphosphate.calciftissueint94:495-509)。
人类间充质干细胞
用人间充质干细胞(msc)测定alp的表达,在saos-2细胞平行测定alp的表达。使用已建立的方法分离和培养细胞(wangxh,etal.themarinesponge-derivedinorganicpolymers,biosilicaandpolyphosphate,asmorphogeneticallyactivematrices/scaffoldsfordifferentiationofhumanmultipotentstromalcells:potentialapplicationin3dprintinganddistractionosteogenesis.marinedrugs12,1131-1147)。
逆转录定量实时pcr分析
应用定量实时rt[逆转录]-pcr(qrt-pcr)技术,来确定圆片对saos-2细胞中以下基因表达水平的影响。简言之,从细胞中提取rna,使用以下引物对进行pcr反应:碳酸酐酶ix(caix;nm_001216人)fwd:5'-acatatctgcactcctgccctc-3'[nt977至nt998](seqidno1)和rev:5'-gcttagcactcagcatcactgtc-3'[nt1105至nt1083](seqidno.2),碱性磷酸酶(alp;nm_000478.4)fwd:5'-tgcagtacgagctgaacaggaaca-3'[nt1141至nt1164](seqidno.3)和rev:5'-tccaccaaatgtgaagacgtggga-3'[nt1418至nt1395](seqidno.4),i型胶原蛋白(coli;nm_000088.3)fwd:5'-gactgccaaagaagccttgcc-3'[nt5073至nt5093](seqidno:5)和rev:5'-ttcctgactctcctccgaaccc-3'[nt51196至nt5175](seqidno:6)和bmp2(骨形态发生蛋白2;nm_001200.2)fwd:5'-accctttgtacgtggacttc-3'[nt1681至nt1700](seqidno:7)和rev:5'-gtggagttcagatgatcagc-3'[nt1785至nt1804](seqidno:no:8)。使用甘油醛-3-磷酸脱氢酶(gapdh)作为参考(nm_002046.5)fwd:5'-ccgtctagaaaaacctgcc-3'[nt929至nt947](seqidno.9)和rev:5'-gccaaattcgttgtcatacc-3'[nt1145至nt1126](seqidno.10)。可以例如在icycler(bio-rad)中,应用各自的icycler软件进行pcr反应。在确定ct值后,计算各个转录物的表达。
制备基于plga的微球
详细描述制备用于动物实验的微球体(wangsf,etal.(2014)bioactiveandbiodegradablesilicabiomaterialforboneregeneration.bone:67:292-304)。使用4mlplga/二氯甲烷溶液(体积比1:5)制备缺少ccp10的微球体;它们被称为“cont-mic”(plga:聚(d,l-丙交酯-共-乙交酯);丙交酯:乙交酯[75:25];mol.wt.66,000-107,000)。对于制备含有caco3/聚p的微球体,以20%的浓度将“ccp10”微球体(“聚p-mic”)加入到plga/二氯甲烷混合物中。通过具有0.8mm孔径的注射器压制粘性反应混合物。通过这种方法,获得平均直径≈820μm的微球体。
如所述测定微球体中聚p的含量(mahadevaiahms,etal.(2007)asimplespectrophotometricdeterminationofphosphateinsugarcanejuices,wateranddetergentsamples.e-journalofchemistry4:467-473)。
测定机械特性
可以例如用配备有已经被熔合到套圈光纤顶部的悬臂的纳米压痕仪,来测定微球体和植入区域(再生区域)的肌肉组织的机械特性(wangsf等人(2014)bioactiveandbiodegradablesilicabiosaterialforboneregeneration.bone67:292-304)。使用该技术来量化降低的杨氏模量(redym)。
体内相容性研究
在实施例中描述的实验中,使用重量在240g和290g(年龄:两个月)之间的(雄性)性别的wistar大鼠;每组使用3只动物。在整个实验期间,随意提供饮食和饮水。在手术前,用10ml/kg体重的环丙沙星处理动物,用于抗生素预防。然后通过肌内注射用氯丙嗪/氯胺酮对动物进行麻醉。接下来在垂直于上肢水平的椎轴的右侧半部和左侧半部,进行≈1cm切口的常规消毒。皮肤切开后,将肌肉切开并分开以容纳微球。将微球体(100μl体积,≈20mg)引入肌肉中,并在更深的层中稳定(vidyas.,parameswarana.,sugumaranvg(1994)comparativeevaluationoftissue.compatibilityofthreerootcanal.sealantsinrattusnorwegicus:ahistopathologicalstudy.endodontology6:7-17)。经过2周、4周或8周的时间,处死动物,切开有周围组织的样本并切片。对样品宏观地检查炎症、感染和变色。将样品固定在福尔马林中,切片,用梅氏苏木精染色,然后通过光学显微镜(例如,用olympusahbt3显微镜)进行分析。
统计分析
使用配对studentt检验对结果进行统计学评估。
序列表
<110>维尔纳·恩斯特·路德维格·格奥尔格·米勒
<120>作为具有形态发生活性的包被层和支架的非晶形无机聚磷酸盐-磷酸钙和碳酸钙颗粒
<130>m32732wo02
<150>gb1420363.2
<151>2014-11-17
<150>gb1513011.5
<151>2015-07-23
<150>gb1515515.3
<151>2015-09-02
<150>gb1518575.4
<151>2015-10-20
<160>10
<170>patentinversion3.5
<210>1
<211>22
<212>dna
<213>智人(homosapiens)
<400>1
acatatctgcactcctgccctc22
<210>2
<211>23
<212>dna
<213>智人
<400>2
gcttagcactcagcatcactgtc23
<210>3
<211>24
<212>dna
<213>智人
<400>3
tgcagtacgagctgaacaggaaca24
<210>4
<211>24
<212>dna
<213>智人
<400>4
tccaccaaatgtgaagacgtggga24
<210>5
<211>21
<212>dna
<213>智人
<400>5
gactgccaaagaagccttgcc21
<210>6
<211>22
<212>dna
<213>智人
<400>6
ttcctgactctcctccgaaccc22
<210>7
<211>20
<212>dna
<213>智人
<400>7
accctttgtacgtggacttc20
<210>8
<211>20
<212>dna
<213>智人
<400>8
gtggagttcagatgatcagc20
<210>9
<211>19
<212>dna
<213>智人
<400>9
ccgtctagaaaaacctgcc19
<210>10
<211>20
<212>dna
<213>智人
<400>10
gccaaattcgttgtcatacc20
- 还没有人留言评论。精彩留言会获得点赞!