一种单靶向还原响应囊泡纳米药物在制备脑肿瘤治疗药物中的应用的制作方法
本发明属于聚合物纳米药物技术领域,具体涉及一种单靶向可穿透血脑屏障并靶向脑肿瘤细胞的还原响应聚合物囊泡载药系统的应用。
背景技术:
脑肿瘤是威胁人类健康的重大疾病。因为病灶部位特殊,并且脑肿瘤具有浸润生长的特点,手术难度大,并且术后会快速复发。如果对脑肿瘤病人进行化疗,血脑屏障的存在又严重阻碍了化疗药物入脑,到达病灶部位。除此之外,给药前扰乱血脑屏障,对脑肿瘤病人实施大剂量的化疗或者放疗还会带来巨大的毒副作用。在过去的几十年,纳米载药系统用于脑肿瘤的治疗成为研究的热点,可是,现有纳米载药系统对小分子抗癌药以及高效低毒的蛋白药物和基因药物的装载效率较低;同时还存在载药纳米系统体内循环不稳定、难以穿透血脑屏障,脑肿瘤细胞摄取低、细胞内药物浓度低等问题;在循环过程中药物被酶降解活性降低,进入癌细胞后不能快速逃离内涵体,导致纳米药物的药效不高,这些都极大的限制了纳米载药系统在脑肿瘤治疗中的应用。此外,即使是利用靶向载药系统进行脑肿瘤治疗,结果也常常不理想。例如,转铁蛋白(Tf)是经典的靶向肿瘤的靶头,利用它构建的脑靶向载药系统非常多,但是因为内源性转铁蛋白的竞争结合,以及在修饰过程中转铁蛋白部分失活,所以在脑肿瘤疾病模型治疗上效果并不理想;而具有双靶向效果的靶向分子修饰的载药脂质体取得的脑肿瘤治疗效果也很有限。考虑到不同靶向分子与血脑屏障和脑胶质瘤细胞表面受体结合能力的差异,研发新的脑肿瘤递药系统显得尤为重要,该载药系统需要同时靶向血脑屏障和胶质瘤细胞,与相关受体亲和能力强,无内源性蛋白竞争结合。
技术实现要素:
本发明的目的是公开一种单靶向还原响应囊泡纳米药物用于脑肿瘤治疗药物的制备,可以高效穿透血脑屏障、深入脑肿瘤实质并且进入脑肿瘤细胞释放药物。本发明用于脑肿瘤的纳米载药系统具备如下几个优点:纳米载药系统包载的药物高效低副作用,即包载的药物对脑肿瘤细胞毒性强,对正常器官和组织毒性低;聚合物纳米系统可以高效包载药物,并且纳米载药系统在血液循环时稳定,在脑肿瘤细胞中可以快速释放药物;纳米载药系统可以高效的穿透血脑屏障,并且被脑肿瘤细胞内吞,然后及时逃离内涵体,在细胞内快速释放药物,同时靶向血脑屏障和胶质瘤细胞,与相关受体亲和能力强,无内源性蛋白竞争结合。
为达到上述发明目的,本发明采用如下技术方案:
单靶向还原响应囊泡纳米药物在制备脑肿瘤治疗药物中的应用。
一种用于脑肿瘤治疗的药物体系,由可逆交联生物可降解聚合物囊泡装载药物得到。
一种脑肿瘤治疗纳米药物,由脑肿瘤治疗药物与分散介质混合得到;所述脑肿瘤治疗药物由可逆交联生物可降解聚合物囊泡装载药物得到。
本发明中,所述单靶向还原响应囊泡纳米药物由可逆交联生物可降解聚合物囊泡装载药物得到;所述单靶向还原响应囊泡纳米药物由可逆交联生物可降解聚合物囊泡装载药物得到;所述可逆交联生物可降解聚合物囊泡由聚合物高聚物自组装后交联得到;所述高聚物为式Ⅰ聚合物、式Ⅱ聚合物的混合物;
式Ⅰ
式Ⅱ
其中R1为靶向分子ApoE,其序列为:Leu Arg Lys Leu Arg Lys Arg Leu Leu Arg Lys Leu Arg Lys Arg Leu Leu Cys;
R2为以下结构式中的一种:
R3为以下结构式中的一种:
R4选自氢或者以下结构式中的一种:
所述式Ⅰ聚合物或者式Ⅱ聚合物中,PEG链段的分子量为3000-8000Da;疏水链段的总分子量为PEG链段分子量的2.5~7倍;疏水链段中PDTC链段的分子量占疏水链段总分子量的10%~30%;PEI的分子量为PEG链段分子量的20%~60%。
本发明中,所述式Ⅰ聚合物或者式Ⅱ聚合物中,DTC与LA/TMC无规共聚形成疏水链段,xy分别表示疏水链段中DTC的重复单元数以及LA/TMC的重复单元数,中括号表示疏水部分为整体,其一端接有亲水PEG;亲水段1为PEG,其分子量为3000-8000Da;疏水段的总分子量为PEG分子量的2.5-7倍;疏水段中PDTC的分子量占整个疏水段总分子量的10%-30%;当亲水段2为PEI时,其分子量为PEG分子量的20%-60%。
本发明中,所述药物为小分子药物、大分子蛋白质药物或基因药物;所述聚乙烯亚胺(PEI)为支化(bPEI)或线性(LPEI),其化学结构式为以下结构式的一种:
、
所述式Ⅰ聚合物或者式Ⅱ聚合物中,PEG链段的分子量为4000-8000Da;疏水链段的总分子量为PEG链段分子量的2.8~6倍;疏水链段中PDTC链段的分子量占疏水链段总分子量的11%~28%;PEI的分子量为PEG链段分子量的20%~50%。
所述式Ⅰ聚合物、式Ⅱ聚合物的物质的量比为(2~20)∶1;所述靶向还原响应囊泡纳米药物中,药物的质量百分数为1%~30%。
本发明中,以高聚物和药物为原料,通过pH梯度法或者溶剂置换法制备单靶向还原响应囊泡纳米药物。
本发明还公开了上述单靶向还原响应囊泡纳米药物在制备穿透血脑屏障药物中的应用,以及上述可逆交联生物可降解聚合物囊泡在制备穿透血脑屏障药物或者脑肿瘤治疗药物中的应用,和上述高聚物在制备穿透血脑屏障药物或者脑肿瘤治疗药物中的应用。
本发明中,当疏水链段包括的PDTC的总分子量为整个疏水链段分子量的10%~30%;所述小分子药物包括阿霉素盐酸盐,大分子蛋白质药物包括皂草素(SAP)、颗粒酶B(GrB),基因药物包括siRNA、mRNA、DNA。
本发明中,所述单靶向还原响应囊泡纳米药物中,药物的质量百分数为1%~30%。本发明的聚合物可自组装形成囊泡,亲水内腔大可高效包载亲水小分子化疗药物,即使载药量达到20wt.%,载药囊泡依然保持稳定,无药物泄露。在聚合物链末端增加修饰PEI或者精胺(Spermine)后,通过静电相互作用和氢键作用可以大大提高囊泡包载大分子药物(蛋白药物或者基因药物)的效率,载药量达到15wt.%时,包封率依然超过80%。同时,上述囊泡在到达癌细胞内后,细胞内的还原性物质GSH又可以快速触发药物释放。此外,本发明的囊泡可以载药穿透血脑屏障进入癌细胞发挥作用。包括脑肿瘤的一系列脑部疾病给药非常困难,不管是大分子药物(蛋白药物和基因药物)还是小分子化疗药物都很难入脑达到有效的治疗浓度。本发明为脑肿瘤的系统给药提供了一种有效方法,较传统纳米载药系统,本发明中的囊泡载药效率、体外稳定性和在肿瘤部位的富集以及药物释放速率都显著提高。
本发明设计的囊泡具有体外以及循环时交联稳定、整个递送过程中保持很高的药物活性、癌细胞内可解交联、同时靶向血脑屏障和脑肿瘤细胞和生物安全性好的特点。囊泡膜的外表面由聚乙二醇(PEG)组成,减少了循环过程中蛋白的吸附,包载大分子药物时,囊泡膜的内表面修饰较低分子量的PEI(600-4800Da)或者精胺,可将大分子药物包载在囊泡内,交联的囊泡膜可保护药物不被降解防止药物泄露,并可延长药物的体内循环时间。囊泡膜为可逆交联的生物可降解且生物相容性好的PTMC或者PLA,侧链的二硫戊环类似人体天然的抗氧化剂硫辛酸,可提供还原敏感的可逆交联,囊泡膜内PEI或者精胺除了用于复合药物如蛋白质、多肽和小分子药物,还能通过质子海绵效应逃逸内涵体,这样的设计不但支持生物药物在血液中的长循环,还可保证在细胞内逃离内涵体,快速解交联,释放药物到靶细胞。囊泡可以载药高效穿透血脑屏障并被脑胶质瘤细胞内吞。
本发明公开的单靶向还原响应囊泡纳米药物的制备方法可举例包括以下步骤:
(1)将PEG-P(TMC-DTC)或者PEG-P(LA-DTC)的末端羟基用羟基活化剂比如氯甲酸对硝基苯酯(NPC)活化,再与PEI反应制得PEG-P(TMC-DTC)-PEI或者PEG-P(LA-DTC)-PEI,或者再与精胺反应制得PEG-P(TMC-DTC)-Sp或者PEG-P(LA-DTC)-Sp;
(2)在PEG-P(TMC-DTC)或者PEG-P(LA-DTC)的PEG端偶联靶向血脑屏障和脑胶质瘤细胞的靶向分子,得到靶向PEG-P(TMC- DTC)或者靶向PEG-P(LA-DTC);
(3)以PEG-P(TMC-DTC)与药物为原料,通过pH梯度法制备抗肿瘤药物;PEG-P(LA-DTC)与药物为原料,通过pH梯度法制备抗肿瘤药物;或者以PEG-P(TMC-DTC)、靶向PEG-P(TMC-DTC)与药物为原料,通过pH梯度法制备抗肿瘤药物;或者以PEG-P(LA-DTC)、靶向PEG-P(LA-DTC)与药物为原料,通过pH梯度法制备抗肿瘤药物;以PEG-P(TMC-DTC)-PEI与药物为原料,通过溶剂置换法制备抗肿瘤药物;PEG-P(LA-DTC)-PEI与药物为原料,通过溶剂置换法制备抗肿瘤药物;以PEG-P(TMC-DTC)-Sp与药物为原料,通过溶剂置换法制备抗肿瘤药物;PEG-P(LA-DTC)-Sp与药物为原料,通过溶剂置换法制备抗肿瘤药物;或者以PEG-P(TMC-DTC)-PEI、靶向PEG-P(TMC-DTC)与药物为原料,通过溶剂置换法制备抗肿瘤药物;或者以PEG-P(LA-DTC)-PEI、靶向PEG-P(LA-DTC)与药物为原料,通过溶剂置换法制备抗肿瘤药物;以PEG-P(TMC-DTC)和靶向PEG-P(TMC-DTC)、药物为原料,或者以PEG-P(LA-DTC)和靶向PEG-P(LA-DTC)、药物为原料,或者PEG-P(TMC-DTC)-PEI和靶向PEG-P(TMC-DTC)、药物为原料,或者以PEG-P(LA-DTC)-PEI和靶向PEG-P(LA-DTC)、药物为原料,或者PEG-P(TMC-DTC)-Sp和靶向PEG-P(TMC-DTC)、药物为原料,或者以PEG-P(LA-DTC)-Sp和靶向PEG-P(LA-DTC)、药物为原料共混自组装、装载药物、交联得到肿瘤主动靶向具有不对称膜结构的药物囊泡,外壳为PEG、靶向分子可介导穿透血脑屏障增加脑胶质瘤细胞的内吞;靶向分子为多肽ApoE。
比如上述制备方法,具体包括以下步骤:
步骤(1)为将PEG-P(TMC-DTC)或者PEG-P(LA-DTC)、羟基活化剂氯甲酸对硝基苯酯NPC溶于干燥的溶剂中反应,然后沉淀、过滤、真空干燥得到活化的PEG-P(TMC-DTC)-NPC或者PEG-P(LA-DTC) -NPC;将PEG-P(TMC-DTC)-NPC或者PEG-P(LA-DTC)-NPC溶液滴加到PEI溶液中反应后,透析、沉淀、抽滤、真空干燥得到PEG-P(TMC -DTC)-PEI或者PEG-P(LA-DTC)-PEI;将PEG-P(TMC-DTC)-NPC或者PEG-P(LA-DTC)-NPC溶液滴加到精胺溶液中反应后,透析、沉淀、抽滤、真空干燥得到PEG-P(TMC-DTC)-Sp或者PEG-P(LA-DTC)-Sp;步骤(2)为将得到聚合物Mal-PEG-P(TMC-DTC)或者Mal-PEG-P(LA-DTC)溶于带有靶向分子的有机溶剂如DMSO中反应得到靶向聚合物;步骤(3)为将原料溶液中加入缓冲溶液中,37摄氏度放置后在相同缓冲溶液中透析,室温孵育交联,得到抗肿瘤纳米药物。本发明可以在加或不加还原剂如二硫代苏糖醇(DTT)和谷胱甘肽(GSH)下室温交联得到可逆交联生物可降解聚合物囊泡。
本发明首次公开了单靶向还原响应囊泡纳米药物在脑肿瘤治疗中的应用,不仅具有制备方法简单、优良的控制释放能力、载体生物相容好、在体内长循环、保护包载药物不被降解的优点,更主要可以高效的穿透血脑屏障进入脑胶质瘤细胞并及时逃离内涵体释放药物,所以该载药囊泡是脑肿瘤治疗的一个有力工具。
附图说明
图1是实施例五的载DOX-HCl囊泡粒径分布图(A)、实施例九的空囊泡对U-87 MG细胞毒性实验(B)和囊泡纳米药物对U-87 MG细胞毒性实验(C);
图2是实施实例十的囊泡纳米药物引起U-87 MG细胞凋亡实验结果;
图3是实施例六的载SAP囊泡的粒径分布图和透射电镜图(A)、穿透血脑屏障体外模型实验(B)、空囊泡对U-87 MG细胞毒性实验(C)和囊泡纳米药物的对U-87 MG细胞毒性实验(D);
图4是实施实例十四的囊泡被U-87 MG细胞内吞及细胞内释放行为结果;
图5是实施例十五荷原位脑胶质瘤小鼠的肿瘤生物荧光(A)、囊泡在荷原位脑胶质瘤小鼠体内分布(B)、囊泡在荷原位脑胶质瘤小鼠主要器官(心、肝脾、肺、肾、荷瘤脑)的分布(C)、囊泡在荷原位脑胶质瘤小鼠的肿瘤荧光强度定量分析(D);
图6是实施例十五囊泡在脑肿瘤区域的分布表征;
图7是实施例十六用囊泡纳米药物治疗后荷原位脑胶质瘤小鼠的体重变化(A)和生存期(B);
图8是实施例十二的空囊泡的细胞毒性实验(A)和载药囊泡细胞毒性实验(B);
图9是实例十八ApoE-PS-siPLK1囊泡的粒径分布图;
图10 是实例十九不同APOE含量的ApoE-PS-siPLK1囊泡在的凝胶电泳结果;
图11 是实例二十ApoE-PS-siRNA囊泡bEnd.3单层细胞模型中评估BBB穿透能力;
图12 是实例二十一ApoE-PS-siCy5囊泡在上室bEnd.3细胞(A)和下室U-87 MG脑胶质瘤细胞(B)内吞的流式细胞仪结果和共聚焦显微镜(CLSM)结果(C);
图13为实施例十九中ApoE-PS-siPLK1囊泡对U-87 MG细胞的PLK1蛋白沉默;
图14为是实施例二十一中ApoE-PS-siCy5囊泡在荷U-87 MG-Luc原位脑胶质瘤小鼠体内的活体成像结果。
具体实施方式
实施例一 PEG5k-P(DTC4.4k-LA19.8k)和ApoE-PEG7.5k-P(DTC 4.4k-LA19.8k)嵌段共聚物的合成
在氮气手套箱内,依次称取MeO-PEG-OH (Mn=5.0 kg/mol, 0.50 g, 100 μmol), LA(2.0 g, 13.9 mmol)和DTC (0.50 g, 2.60 mmol)并溶解在二氯甲烷(DCM,7.0 mL)中,搅拌加入催化剂磷酸二苯酯(DPP,DPP/OH 摩尔比为10/1)。密闭反应器密封好放置40℃油浴中磁力搅拌下反应2天。三乙胺终止反应后在冰乙醚中沉淀两次、抽滤、常温真空干燥后得到PEG5k-P(DTC4.4k-LA19.8k)。
ApoE-PEG7.5k-P(DTC4.4k-LA19.8k)的合成分两步,第一步与PEG5k-P(DTC4.4k-LA19.8k)的合成类似,用Mal-PEG-OH (Mn=7.5 kg/mol)替代MeO-PEG-OH (Mn=5.0 kg/mol)引发DTC和LA的开环聚合反应得到Mal-PEG7.5k-P(DTC4.4k-LA19.8k)。后者和多肽ApoE(其序列为:Leu Arg Lys Leu Arg Lys Arg Leu Leu Arg Lys Leu Arg Lys Arg Leu Leu Cys)按照摩尔比为1:1.2反应:在氮气下将DMSO中溶解的ApoE滴加到DMSO溶解的Mal-PEG7.5k- P(DTC4.4k-LA19.8k)中,37度搅拌反应8小时。用DMSO透析24时再用水透析12时后,冷冻干燥得到ApoE-PEG7.5k-P(DTC4.4k-LA19.8k)。通过核磁和BCA法表征多肽ApoE的接枝率约为96%。
实施例二 合成嵌段共聚物PEG5k-P(DTC2k-TMC15k)和PEG5k- P(DTC2k-TMC15k)-bPEI1.8k
在氮气手套箱内,依次称取MeO-PEG-OH (Mn=5.0 kg/mol, 0.50 g, 100 μmol), TMC (1.52 g, 14.55 mmol) 和DTC (0.23 g, 1.18 mmol) 并溶解在二氯甲烷(DCM,7.0 mL)中,搅拌加入催化剂磷酸二苯酯(DPP,DPP/OH 摩尔比为10/1)。密闭反应器密封好放置40℃油浴中磁力搅拌下反应2天。三乙胺终止、冰乙醚中沉淀两次、抽滤、真空干燥后得到PEG5k-P(DTC2k-TMC15k)。
PEG5k-P(DTC2k-TMC15k)的末端羟基氯甲酸对硝基苯酯NPC活化,再与支化PEI(bPEI)的伯胺反应制得。具体的,PEG5k-P(DTC2k-TMC15k) (0.4 g, 羟基0.017 mmol)和NPC(50 mg, 0.09 mmol)溶于干燥的DCM中在0℃下反应24小时,然后在冰乙醚中沉淀、过滤、真空干燥得到PEG5k-P(DTC2k-TMC15k)-NPC。然后将产物溶于3 mL DCM后滴加到3 mL溶有bPEI (Mn=1.8 kg/mol) (235 mg, 0.13mmol)的DCM中,30℃下反应24小时后,在DCM和甲醇(体积比为1:1)中透析(MWCO 7000) 48小时,接着在冰乙醚中沉淀两次、抽滤并室温真空干燥得到产物PEG5k-P(DTC2k-TMC15k)-bPEI1.8k。产率:93.4%。1H NMR (400 MHz, DTCl3):PEG: δ 3.38, 3.65; TMC: δ 4.24, 2.05; DTC: δ 4.32, 3.02, PEI: δ 2.56-2.98。通过积分可知,聚合物的分子量和设计的理论分子量吻合,且GPC测定分子量分布窄,均说明该反应活性可控。
实施例三 靶向共聚物的合成
靶向聚合物的合成有多种方式,取决于PEG的端功能化基团。ANG-PEG7.5k-P(DTC2k-TMC15k)的合成分两步。第一步与实施例一中PEG5k-P(DTC2k-TMC15k)的合成类似,但用Mal-PEG-OH(Mn=7.5 kg/mol)替代MeO-PEG-OH(Mn=5.0 kg/mol)做引发剂,引发DTC和TMC的开环聚合得到Mal-PEG7.5k-P(DTC2k-TMC15k)。第二步后者和多肽ApoE的巯基按摩尔比1:1.2发生迈克尔加成反应。在氮气下将靶向多肽ApoE的DMSO溶液解滴加到Mal-PEG7.5k-P(DTC2k-TMC15k)的DMSO溶液中,37度搅拌反应8小时后在DMSO透析24时再用二次水透析12时,冷冻干燥得产物ApoE -PEG7.5k-P(DTC2k-TMC15k),产率92%。核磁积分可知聚合物分子量为7.5-(2.0-14.7) kg/mol。核磁和BCA法表征ApoE的接枝率为93%。
Mal-PEG7.5k-P(DTC2k-TMC15k)如同实施例二第二步一样,端羟基活化、与PEI反应得到Mal-PEG7.5k-P(DTC2k-TMC15k)-bPEI1.8k,后者再与多肽ApoE的巯基室温下加成反应,得到靶向聚合物ApoE-PEG7.5k-P(DTC2k-TMC15k)-bPEI1.8k。
实施例四 合成嵌段聚合物PEG5k-P(TMC15k-DTC2k)-Sp
同实施例二法合成的PEG5k-P(DTC2k-TMC15k)-NPC溶于3 mL DCM后,滴加到3 mL溶有精胺(26 mg,0.13mmol)的DCM中,30℃下反应48小时后,在DCM和甲醇(体积比为1:1)中透析(MWCO 7000)48小时、冰乙醚沉淀、抽滤、真空干燥得到PEG5k-P(DTC2k-TMC15k)-Sp。产率:94.7%。核磁和TNBSA法表征Sp的接枝率为97%。表1列出了各聚合物制备条件和产物的核磁表征结果,通过连接基团可接上靶向分子ApoE。
表1 各个聚合物制备条件和产物的核磁表征结果
实施例五 制备载DOX•HCl、以ApoE为靶向分子的交联囊泡
将PEG5k-P(DTC2k-LA15k)和ApoE-PEG7.5k-P(DTC2k-LA15k)分别溶于DMF(10mg/mL)。按ApoE-PEG7.5k-P(DTC2k-LA15k)和PEG5k-P(DTC2k-LA15k)物质的量比1:4把100μL聚合物溶液滴入950μL匀速搅拌的柠檬酸缓冲溶液(5 mM,pH 4.0)中,加入磷酸氢二钠饱和溶液将pH调至7.8,快速加入相应体积的阿霉素盐酸盐溶液(5mg/mL),继续搅拌10min,37度静置交联12h,用磷酸盐缓冲溶液(10mM,pH 7.4)透析(MWCO 7,000)8h,每2h换一次缓冲溶液即得到载DOX•HCl的囊泡ApoE-PS-DOX。PEG5k-P(DTC2k-LA15k用同样的方法可得到无靶向的载DOX•HCl囊泡PS-DOX。图1A和表2显示载不同比例DOX•HCl(10-20wt%)的交联囊泡粒径为78-111 nm,粒径分布为0.11-0.16。紫外分光光度计测定DOX•HCl的包载效率为37.4%-55.5%。
表2 载DOX•HCl囊泡的表征结果
实施例六 制备载SAP的交联囊泡和以ApoE为靶向分子的交联囊泡
将PEG5k-P(DTC2k-TMC15k)-bPEI1.8k和靶向聚合物ApoE-PEG7.5k -P(DTC2k -TMC15k)分别溶于DMSO(10mg/mL)。按物质的量比4:1把100μL聚合物溶液注入950 μL含不同浓度SAP的HEPES(5 mM,pH 6.8)缓冲溶液中,37度静置,交联过夜。将得到溶液在PB中(10 mM, pH 7.4)透析(MWCO 350,000)即得到载SAP的囊泡ApoE-PS-SAP。将聚合物替换为PEG5k-P(DTC2k-TMC15k)-bPEI1.8k,采用同样的方法可得到无靶向的载SAP 囊泡PS-SAP。靶向聚合物占总聚合物摩尔比分别为0、10%、20%、30%,对应囊泡表示为PS、ApoE10-PS、ApoE20-PS、ApoE 30-PS。图3A和表3显示载不同比例SAP(5%-15wt%)的交联囊泡粒径为77-86nm,粒径分布为0.11-0.16,透射电镜图(图3A)也可以清晰的看到囊泡的中空结构。BCA法测定SAP的包载效率为73.2%-91.8%。
表3 载SAP囊泡的表征结果
aSAP载药量由BCA法测定;b室温PB (pH 7.4, 10 mM) 中测定;c室温PB中测定
实施例七 制备载GrB的交联囊泡和以ApoE为靶向分子的交联囊泡
PEG5k-P(DTC2k-TMC15k)-Sp和ApoE-PEG7.5k-P(DTC2k-TMC15k)制备囊泡装载颗粒酶B(GrB)同实施例六,得到载GrB的囊泡ApoE-RCCP-GrB和无靶向的载GrB囊泡RCCP-GrB(表4),结合图8A显示载颗粒酶B的交联囊泡粒径为77-86 nm,粒径分布PDI为0.08-0.16。
a BCA法测定SAP载药量;b室温PB (pH 7.4, 10 mM) 中测定;c室温PB中测定
实施例八 制备载SAP的交联囊泡和以ANG为靶向分子的交联囊泡
PEG5k-P(DTC2k-TMC15k)-bPEI1.8k和ANG-PEG7.5k-P(DTC2k- TMC15k)制备囊泡装载蛋白质SAP同实施例六,得到载SAP的囊泡ANG-PS-SAP和无靶向的载SAP囊泡PS-SAP。表5测试显示载不同比例SAP(5%-10 wt%)的交联囊泡粒径为68-88 nm,粒径分布为0.08-0.15。BCA法测定SAP的包载效率为81.3%-92.5%。
表5 载SAP囊泡ANG-PS-SAP和PS-SAP的表征结果
aSAP载药量由BCA法测定;b 粒径于室温,PB缓冲液(pH 7.4, 10 mM) 中测定
实施例九 MTT法测试空囊泡和载DOX-HCl交联囊泡对U-87 MG的细胞毒性
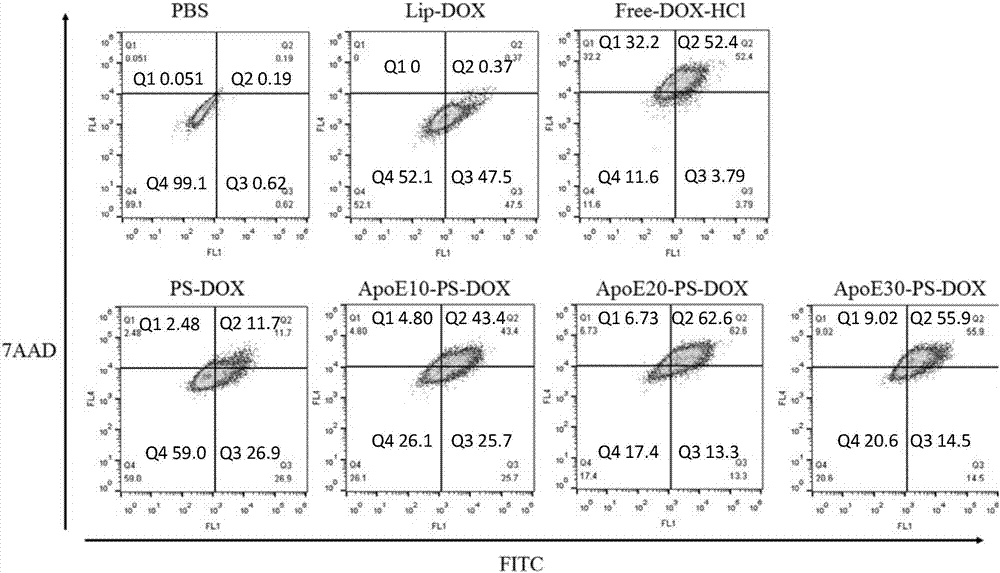
用MTT实验来评估实施例四中制备的囊泡的细胞毒性结果表明,ApoE-PS-DOX对LRP-1、LRP-2和LDLR过表达的U-87 MG 细胞的毒性很高(图1C),而无靶向组PS-DOX和Lipo-DOX在相同药物浓度下,细胞毒性明显低。表明ApoE-PS-DOX能够特异性结合相关受体并高效进入U-87MG。另外,靶向分子密度对靶向能力又有很大影响:20%的ApoE表现出最佳靶向能力。空载体的生物相容性良好,浓度达到1mg/mL时细胞存活率依然在95%以上(图1B)。
实施例十 流式细胞仪来评价载DOX囊泡的细胞内吞和引起细胞凋亡的能力
用流式细胞仪来评价实施例四制备的囊泡被U-87 MG细胞内吞和诱导其凋亡能力。载DOX的囊泡在U-87 MG细胞中有少量内吞,载DOX的ApoE-PS-DOX囊泡的细胞内吞量明显增加,20%的ApoE表现最佳。图2流式凋亡实验结果显示ApoE-PS-DOX引起的细胞凋亡明显多于PS-DOX以及Lipo-DOX,20%的ApoE囊泡引起了更多的细胞凋亡,与DOX-HCl引起的凋亡相近。
实施例十一 MTT法测试PS-SAP和ApoE-PS-SAP对U-87 MG的细胞毒性
用MTT实验来评估实施例六制备的载SAP囊泡的抗癌活性(图3A),自由 SAP在药物浓度达到40 nM时,细胞存活率依然高于90%,而PS-SAP明显提高了SAP的细胞毒性,细胞存活率下降至70%,而ApoE-PS-SAP对LRP-1过表达的U-87 MG 细胞具有更强的细胞毒性,其IC50值只有10.2 nM。上述结果表明修饰靶向分子ApoE可以近一步提高载药囊泡的细胞内吞效率,提高药物的细胞毒性。同时,靶向和非靶向的空载体却都显示出很好的生物相容性(图3C、D)。
实施例八制备的载SAP囊泡ANG-PS-SAP对于U-87 MG 细胞的IC50值为 30.2 nM。结果说明不同的脑肿瘤靶向会有不同的结果。
实施例十二 MTT法测试RCCP-GrB和ApoE-RCCP-GrB对U-87 MG的细胞毒性
用MTT实验来评价空囊泡的生物相容性以及实施例七制备的载颗粒酶B囊泡的抗癌活性。在空囊泡浓度达到0.4mg/mL时,细胞存活率依然高于95%,显示出载体有良好的生物相容性(图8B)。ApoE-RCCP-GrB对U-87 MG 细胞具有很强的细胞毒性,其IC50值为4 nM,而无靶向组RCCP-SAP和free SAP在药物浓度达到100 nM时,细胞存活率依然高于70%和90%(图8C)。上述结果表明ApoE-RCCP-GrB能够通过高效进入U-87 MG,且在细胞内快速释放蛋白药物。
实施例十三 交联囊泡穿透血脑屏障体外模型评价
用BBB的体外模型来研究Cy5标记的载药囊泡穿透血脑屏障的效率。首先将bEnd.3细胞 (1 × 105细胞/孔) 铺于24孔板的上室内,下室加入800 μL DMEM培养基孵育48小时后,通过跨内皮电阻(TEER)仪(World Precision Instruments)测量bEnd.3单层的紧密度。其次,将培养液更换为无FBS 的DMEM,当bEnd.3细胞单层TEER值超过200 Ω.cm2时,50 μL HEPES的不同ANG密度的囊泡加入transwell上室,37 ℃、50 rpm的摇床中孵育24小时收集下室或上室培养基,并用等体积的新鲜培养基代替。每收集一次都对TEER进行监测。通过荧光分光光度计(Thermo Scientific)测量外流比率。结果表明,加入transwell孵育24小时后,ApoE20-PS-Cy5显示出更高的穿透效率(26.7%),明显高于非靶向对照组PS-Cy5(6.1%)和 ANG20-PS-Cy5(13.6%)和ANG30-PS-Cy5(11.7%)(图3B)。结果说明ApoE穿透血脑屏障的效率很高,约为ANG多肽修饰囊泡的两倍。
实施例十四 囊泡纳米药物的U-87 MG细胞内吞和细胞内释放
用FITC标记的细胞色素C(FITC-CC)作为模型蛋白装载到囊泡中考察囊泡纳米药物ApoE20-PS-FITC-CC的细胞内吞和胞内释放行为。向24空板中的U-87 MG细胞(5000个)加入载FITC-CC的囊泡纳米药物(FITC-CC浓度为50 nM)孵育4 h后,更换为纯培养基继续培养4 h。依次用罗丹明B染细胞骨架30分钟、DAPI染核10分钟,每次染色后用PBS洗涤三次。然后用共聚焦荧光显微镜观察,发现ApoE20-PS-FITC-CC被细胞大量内吞,明显高于无靶向PS-FITC-CC,而FITC-CC则不能被细胞内吞(图4)。
实施例十五 交联囊泡在荷原位脑胶质瘤小鼠体内生物分布和在体内穿透肿瘤区域脑微血管能力的考察
所有动物实验操作均在苏州大学动物中心及动物保护及使用委员会批准下进行。活体成像系统就被用来考察不同靶向密度的囊泡在肿瘤部位富集能力的差异。肿瘤细胞的生物荧光可以清楚显示肿瘤的位置和相对大小(图5A),图5B是尾静脉注射不同ApoE靶向分子密度、载Cy5标记的细胞色素C(Cy5-CC)的囊泡在24h时在小鼠体内的分布情况。囊泡选择性的富集在脑肿瘤部位,正常脑组织几乎观察不到载Cy5-CC囊泡的荧光。注射纳米药物24h后,将荷瘤鼠的脑取出后发现,囊泡选择性的富集在脑肿瘤部位这与活体观察到的结果一致(图5C)。脑肿瘤部位的荧光强度定量分析发现ApoE20-PS显示出最好的富集效果,分别是ANG10-PS和ANG30-PS的1.9倍和1.2倍(图5D)。载Cy5-CC的囊泡ApoE20-PS-Cy5-CC 穿透肿瘤与正常组织边界的血管进入肿瘤实质,高效富集在肿瘤(图6)。这与BBB体外模型结果一致,证明ApoE可高效介导囊泡纳米药物穿过血脑屏障富集在肿瘤实质。
实施例十六 载蛋白质药物交联囊泡对荷原位脑胶质瘤小鼠的治疗
原位荷脑胶质瘤鼠被用来评估载蛋白囊泡的体内抗肿瘤效果,肿瘤的生物荧光被用来检测肿瘤的大小。原位脑胶质瘤模型的建立:将U-87 MG-Luc细胞(1×107细胞悬浮于50 μL的0.9%NaCl中)注射到BALB/c载体裸鼠的侧腹。当其肿瘤体积增长至约300 mm 3时,将载体小鼠处死以收获皮下肿瘤。然后将约2 mg切碎的脑肿瘤组织用专门制作的螺旋桨植入到每只麻醉动物的左侧纹状体(前颅侧2 mm,深3 mm)中(使用24#套管针腹膜内注射戊巴比妥钠,剂量80 mg/kg)。由IVIS Lumina系统观察肿瘤生长情况,在成像前10-15分钟,腹腔注射100 μL荧光素酶(150 mg/kg)为底物。约两周后开始实验。图7A显示连续尾静脉给药载SAP囊泡后,无靶向组PS-SAP有一定的抑制作用,ApoE-PS-SAP组显示出更好的肿瘤抑制效果。随着脑胶质瘤的恶化,到接种后第19天,PBS组小鼠状态变差,并且有小鼠死亡。PS-SAP组虽然有一定的抗肿瘤效果,但还是有明显的体重下降,并且在接种后第26天也出现动物死亡。而ApoE-PS-SAP组直到45天才开始有小鼠死亡。不同组的中位生存期(图7B)分别为20 天 (PBS) 、21 天 (SAP) 、29天(PS-SAP(0.25mg/kg))、33天(PS-SAP(0.5mg/kg))、51天(ApoE-PS-SAP(0.25 mg/kg))、58天 (ApoE-PS-SAP(0.5mg/kg))。
类似地,尾静脉给药载GrB囊泡实验中,PBS组接种后第18天开始有小鼠死亡;无靶向组PS-GrB有一定的抑制作用,但体重下降明显,第27天出现小鼠死亡。ApoE-PS-GrB组显示出更好的抑瘤效果:50天才开始有小鼠死亡。中位生存期分别为20天(PBS)、21天(GrB)、31天(PS-GrB,0.05mg/kg)、35天(PS-GrB,0.1mg/kg)、56天(ApoE-PS-GrB,0.05 mg/kg)和65天(ApoE-PS-GrB,0.1 mg/kg)。
实施例十七 载DOX、ApoE为靶向分子的囊泡治疗荷原位脑胶质瘤小鼠
如实施例五制备的装载DOX•HCl、基于PEG5k-P(DTC2k-LA15k)和ApoE-PEG7.5k-P(DTC2k-LA15k)的ApoE20-PS-DOX尾静脉给药。PBS组接种后第18天开始有小鼠死亡;PS-DOX有一定的抑制作用,但体重下降明显,第28天出现小鼠死亡。里葆多组动物瘦弱,毒性明显,21天开始有死亡。ApoE-PS-DOX组显示出更好的抑瘤效果:50天才开始有小鼠死亡。中位生存期分别为20天(PBS)、24天(里葆多,6 mg DOX/kg)、31天(PS-DOX,10mg DOX/kg)和56天(ApoE-PS-DOX,10mg DOX/kg)。
实施例十八 制备装载各种siRNA的囊泡和靶向囊泡
通过溶剂交换法复合装载各种siRNA,包括特异性的siPLK1、荧光标记的siRNA(Cy5-siRNA)和非特异性siRNA(siScramble)。100 μL溶于DMSO的聚合物 PEG5k-P(DTC2k-TMC15k)-Spermine或者和特定比例的靶向聚合物ApoE-PEG7.5k-P(DTC2k-TMC15k) ;或是PEG5k-P(DTC2k-TMC15k)-Sp或者和特定比例的ApoE-PEG7.5k-P(DTC2k-TMC15k)打入900 μL含有特定比例siRNA缓冲溶液(1 mg/mL)的HEPES (5 mM, pH 6.8)中,室温搅拌5分钟、摇床25 ℃、100 rpm交联过夜,HEPES中透析得到各种载siRNA的囊泡。DLS结果(图9)显示粒径为40-50 nm,载10 wt.% siRNA的ApoE-PS粒径为44 nm,粒径分布为0.13。表6为PS-siPLK1和ApoE-PS-siPLK1、ApoE-PS-siScramble的粒径与装载效率。
表6 ApoE-PS-siRNA的粒径与包载效率
实施例十九 ApoE-PS-siPLK1的凝胶电泳分析
琼脂糖胶中分别加入20 μL的2.5% ApoE-PS-siPLK1,5% ApoE-PS-siPLK1,7.5% ApoE-PS-siPLK1和10% ApoE-PS-siPLK1,自由siRNA,以及用10 mM GSH处理过夜后的2.5% ApoE-PS-siPLK1,5% ApoE-PS-siPLK1,7.5% ApoE-PS-siPLK1和10% ApoE-PS-siPLK1,自由siRNA,在TBE电泳缓冲液中跑胶(100 V, 30 min)后,由Molecular Imager FX(Bio-Rad, Hercules,Ex/Em: 532/605 nm)拍照凝胶图片,通过Quantity One 软件(Bio-Rad)分析,见图10,琼脂糖凝胶阻留法表明,ApoE-PS可以完全、紧实包裹siRNA,证明ApoE-PS-siRNA稳定性优异。在10 mM GSH存在下过夜孵育,囊泡的解交联,大部分siRNA释放出来。
实施例二十 ApoE-PS-siCy5(siCy5: Cy5-siRNA)穿透血脑屏障实验
如实施例十三建立体外BBB模型。当bEnd.3细胞单层TEER值超过200 Ω.cm2时,50 μL HEPES的载Cy5-siRNA的囊泡(ApoE-PS-siCy5或PS-siCy5)加入上室。 然后37 ℃、50 rpm的摇床中孵育6、12或24小时。图11是表明ApoE-PS-siRNA具有显著的BBB穿透能力。
实施例二十一 ApoE-PS-siCy5流式细胞仪及共聚焦显微镜实验
ApoE-PS-siCy5和PS-siCy5首先穿透血脑屏障随后被脑胶质瘤细胞U-87 MG的内吞及释放行为通过流式细胞仪及CLSM检测。如实施例十三建立体外BBB模型。首先将bEnd.3细胞 (1×105细胞/孔) 铺于24孔板的上室内,下室加入800 μL DMEM培养基孵育24小时,把下室培养基移去,加入U-87 MG 细胞(2×105细胞/孔)继续孵育24小时。当bEnd.3细胞单层TEER值超过200 Ω.cm2时,50 μL HEPES的载Cy5-siRNA的囊泡(ApoE-PS-siCy5或PS-siCy5)加入上室,然后摇床孵育24小时。随后分别收集上室bEnd.3细胞和下室U-87 MG细胞,流式细胞仪检测。图12分别为ApoE-PS-siCy5在bEnd.3细胞(A)和U-87 MG细胞(B)的内吞的流式结果,说明ApoE-PS-siCy5能被有效内吞进入细胞。图12C显示ApoE-PS-siCy5孵育细胞荧光强度明显强于PS-siCy5。MTT实验表明ApoE-PS空囊泡在浓度高达0.5 mg/mL时也没有毒性(细胞存活率>88%),佐证了本发明的囊泡优异的生物相容性。
实施例二十二qRT-PCR定量ApoE-PS-siPLK1的体外基因沉默能力
按实施例十八制备装载治疗性基因siRNA(siPLK1)的囊泡ApoE-PS-siPLK1。用实时荧光定量基因扩增荧光检测系统(qRT-PCR)研究ApoE-PS-siPLK1内源性基因沉默活性实验,类似球激酶(PLK1)作为靶向基因。U-87 MG细胞悬浮于含有10% FBS的DMEM培养基中铺于6孔板(3×105 个细胞/孔)培养24 h后,分别加入100 µL ApoE-PS-siPLK1、ApoE-PS-siScramble和PS-siPLK1 (最终siRNA浓度为200 nM和400 nM)孵育48 h。细胞经PBS清洗并收集PLK1RNA,反转并由qPCR测试得到(GAPDH作为内参基因)。图13可见,ApoE-PS-siPLK1组的PLK1 mRNA表达量与显著低于PS-siPLK1和ApoE-PS-siScramble,证明其靶向性及序列特异性基因沉默能力。另外,在蛋白水平上进一步验证了ApoE-PS-siPLK1在U-87 MG细胞中序列特异性沉默PLK1蛋白的能力。本文发明的ApoE-PS囊泡能有效包裹siRNA,有效被细胞内吞,通过PEI质子海绵效应逃离内涵体,细胞质还原环境下快速释放siRNA,高效沉默相应基因。
实施例二十三 ApoE-PS-siGL3的体内基因沉默
如实施例十六建立U-87 MG-Luc原位脑胶质瘤肿瘤。约两周后开始实验,分别尾静脉注射200 μL HEPES的ApoE-PS-siGL3 和ApoE-PS-siScramble (20 μg siRNA/鼠)。原位脑胶质瘤裸鼠的脑部荧光在ApoE-PS-siGL3给药前后发生显著变化;脑部生物荧光的定量分析发现,注射ApoE-PS-siGL3的24及48 h后,脑部生物荧光强度分别降低59%及79%,证明ApoE-PS-siGL3诱导脑组织荧光素酶基因有效表达,没有观察到ApoE-PS-siScramble小鼠脑部荧光强度的变化,证实特异序列能致使生物荧光基因沉默。
实施例二十四 ApoE-PS-siCy5体内活体成像
荷原位脑胶质瘤U-87 MG-Luc裸鼠随机分为两组,分别尾静脉注射200 μL HEPES的ApoE-PS-siCy5 和PS-siCy5 (20 μg Cy5-siRNA/鼠)。在2、4、8、12和24小时,小鼠通过异氟烷麻醉、近红外荧光成像系统(Lumina, IVIS II)获取荧光图(Ex.633 nm,Em.670 nm)。在图片获取过程中,由小动物麻醉机麻醉小鼠。通过Lumina II软件拍摄并分析图片。图14为肿瘤部位Cy5-siRNA荧光图,显示ApoE-PS-siCy5组小鼠在注射2 h后,观察到肿瘤部位Cy5-siRNA荧光很强;PS-siCy5在肿瘤部位积累量显著减少。结果表明主动靶向在肿瘤高富集及久持续上发挥重要作用。
实施例二十五 荷U-87 MG-Luc原位脑肿瘤裸鼠的治疗实验
如实施例十六建立原位U-87MG-Luc胶质瘤模型。接种时定位第0天,约10 d后肿瘤荧光强度达到106时开始治疗。小鼠称重并随机分为4组(每组8只):ApoE-PS-siPLK1、PS-siPLK1、ApoE-PS-siScramble和PBS。小鼠经尾静脉每两天注射一次,剂量为60 µg siRNA/鼠。小鼠的相对体重以它们初始体重为标准。第20天治疗终止,每组任意取一只小鼠处死,取出主要器官清洗。之后,浸泡在4%的福尔马林并包埋于石蜡中,由H&E染色并由正置显微镜拍照(Olympus BX41)。40天内观察各组的生存曲线(每组7只)。荧光成像跟踪肿瘤生长情况结果表明,和PBS组相比,PS-siPLK1能部分抑制肿瘤增长,而ApoE-PS-siPLK1显著抑制肿瘤增长。ApoE-PS-siScrambl和PBS组小鼠情况类似,肿瘤快速增长。脑部荧光定量分析显示了ApoE-PS-siPLK1的高效肿瘤抑制能力要显著强于PS-siPLK1;ApoE-PS-siPLK1组小鼠体重几乎无变化,而PS-siPLK1、ApoE-PS-siScramble及PBS组小鼠体重有所降低。生存曲线显示ApoE-PS-siPLK1组小鼠生存期明显延长。ApoE-PS-siPLK1、PS-siPLK1、ApoE-PS-siScramble和PBS组小鼠生存中值分别为50、34.0、25.0及21.0天。肿瘤的组织学分析表明ApoE-PS-siPLK1引发了大面积脑肿瘤细胞的凋亡,但对心、肝、脾、肺及肾基本无伤害。这些结果表明ApoE-PS-siPLK1介导安全、高效、靶向递送siRNA至荷原位脑肿瘤小鼠。
序列表
<110> 苏州大学
<120> 一种单靶向还原响应囊泡纳米药物在制备脑肿瘤治疗药物中的应用
<160> 1
<170> SIPOSequenceListing 1.0
<210> 1
<211> 18
<212> PRT
<213> 人工合成(Artificial)
<400> 1
Leu Arg Lys Leu Arg Lys Arg Leu Leu Arg Lys Leu Arg Lys Arg Leu
1 5 10 15
Leu Cys
- 还没有人留言评论。精彩留言会获得点赞!