一种补肾舒脊的中药组合物的质量检测方法及其应用与流程
本发明涉及中药制剂领域,具体涉及一种补肾舒脊的中药组合物的质量检测方法及其应用。
背景技术:
强直性脊柱炎(ankylosingspondylitis,as)是一种原因未明的慢性进行性免疫炎性疾病,主要侵犯中轴关节如骶髂关节、脊柱骨突、脊柱旁软组织及外周关节,并可伴发关节外表现。临床主要表现为腰骶、脊背、颈项、臀、髋部疼痛以及关节肿痛,严重者可发生脊柱畸形和关节强直,影响患者的身心健康,并波及眼、心、肺、肾等脏器,致残率较高。
国内指南的治疗原则认为强制性脊柱炎目前尚无根治方法,如果患者能够得到及早发现及时诊断及合理治疗,可以控制症状并改善预后,对于晚期患者,必要时可行外科手术,矫正畸形关节,从而改善和提高患者生活质量及预后。治疗强直性脊柱炎的传统西医药物有非甾体抗炎药(nsaids),改善病情抗风湿药(dmards)和糖皮质激素及生物制剂等。由于as病因未明,目前西医尚缺乏理想的特效治疗药物。大量的临床经验表明,中医药治疗as显示出了独特的疗效。有从分期入手治疗者,有强调辨证分型治疗者,有些为单方单法治疗,有辅以针灸及外治法等治疗,但基本上认为本病正虚邪实,多采用补益肝肾、清热除湿、祛风散寒、活血祛瘀、通络止痛等治法。
鉴于中药是一个由多种成分构成的复杂体系,且其成分受多个因素影响,如品种、产地、采收、炮制加工等,而中药中多样性、复杂性的成分是其疗效良好、作用广泛、副作用较小的物质基础,该物质基础长期处于一种模糊、难以全面评价的状态。经多味药味的配伍煎煮后,复方的质量更是无法用明确、有效、合理的指标进行评价,然而中药复方治疗效果与其质量有着直接关系,中药复方质量不良会直接导致治疗效果的不佳,这需要对中药复方进行质量控制以保证疗效的稳定性、可控性。中药质量的控制需要能对全部药效成分进行监控,建立的质量控制体系才能保证用药的有效性、可控性、安全性。因此,建立中医药基础理论特色的现代质量管控体系,解决中药分析的难题,改进现有质量控制方法是当今研究的热点。
技术实现要素:
因此,本发明要解决的技术问题在于提供一种补肾舒脊的中药组合物的质量检测方法及其应用,克服了因中药药味繁多、成分复杂且相互干扰以致不能全面的、清晰地对所述中药组合物的质量进行检测监控的缺陷,提高了所述中药组合物质量的稳定性、一致性和可控性。
本发明提供了一种补肾舒脊的中药组合物的质量检测方法,所述中药组合物包括如下重量份的原料药组成:骨碎补8-12份、狗脊4-8份、杜仲4-8份、桂枝4-8份、鹿角2-6份、延胡索5-9份、续断5-9份、秦艽5-9份、青风藤7-11份和羌活4-8份;
所述中药组合物的质量检测方法包括如下含量测定步骤:
a、柚皮苷的含量测定
供试品溶液的制备:取所述中药组合物,加入甲醇溶液,超声提取,过滤,滤液蒸干,加水溶解,柱层析提纯,收集洗脱液蒸干,加甲醇溶液溶解定容,得供试品溶液;
对照品溶液的制备:取柚皮苷对照品,加入有机溶剂制成对照品溶液;
色谱条件与系统适用性试验:照高效液相色谱法测定,以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.1vt%磷酸溶液为流动相b,按照流动相a:流动相b的体积比为10-30%:90-70%;检测波长为260-300nm,理论塔板数按柚皮苷计算应不低于2000;或
照高效液相色谱法测定,以氨基键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,按照流动相a:流动相b的体积比为70-100%:30-0%;检测波长为260-300nm,理论塔板数按柚皮苷计算应不低于2000;
测定法:分别吸取对照品溶液和供试品溶液,注入液相色谱仪,测定并计算。
进一步地,在所述柚皮苷的含量测定中,所述供试品溶液制备的具体方法为:取所述中药组合物,精密称定,精密加入70-100vt%甲醇溶液10-100ml(vt%为体积浓度),超声处理0.1-1.0h,取出,放冷,再称定重量,用70-100vt%甲醇溶液补足减失的重量,摇匀,滤过,取滤液5-50ml,蒸干,残渣加水1-10ml,溶解,放冷,过聚酰胺色谱柱,以10-100ml水液洗脱,弃去水液,再用甲醇10-100ml洗脱,收集洗脱液蒸干,再用甲醇定容至1-50ml,摇匀,滤过,即得。
优选地,所述色谱条件与系统适用性试验的具体方法为:照高效液相色谱法测定,以氨基键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,按照流动相a:流动相b的体积比为90%:10%;检测波长为283nm,理论塔板数按柚皮苷计算应不低于2000;
或,色谱条件与系统适用性试验的具体方法为:照高效液相色谱法测定,以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.1vt%磷酸溶液为流动相b,按照流动相a:流动相b的体积比为20%:80%;检测波长为283nm;理论塔板数按柚皮苷计算应不低于2000。
进一步地,所述中药组合物的质量检测方法还包括如下含量测定方法或鉴别方法中的至少一种:
b、青藤碱的含量测定
供试品溶液的制备:取适量所述中药组合物,精密称定,精密加入体积浓度40-95%乙醇溶液10-50ml,超声处理0.1-1.0h,放冷,用体积浓度为40-95%乙醇溶液补足减失的重量,摇匀,滤过,取续滤液,即得;
对照品溶液的制备:取青藤碱对照品适量,精密称定,加体积浓度50-100%甲醇溶液制成青藤碱对照品溶液,即得;
色谱条件与系统适用性试验:照高效液相色谱法测定,以十八烷基硅烷键合硅胶为填充剂;以甲醇为流动相a,以ph9.0的磷酸盐缓冲液为流动相b,流动相a:流动相b的体积比为25-40%:75-60%;检测波长为240-280nm;理论塔板数按青藤碱计算应不低于1500;
测定法:分别精密吸取对照品溶液与供试品溶液各2-20μl,注入液相色谱仪,测定并计算;或
c、延胡索的鉴别:
供试品溶液的制备:取所述中药组合物0.5-4g,加体积浓度90-100%的甲醇溶液30-80ml,超声处理15-60分钟,滤过,滤液蒸干,残渣加水5-20ml使溶解,加浓氨试液调至碱性,用乙醚振摇提取1-5次,每次5-20ml,合并乙醚液,蒸干,残渣加体积浓度90-100%的甲醇1-3ml使溶解,即得;
对照品药材溶液和对照品溶液的制备:取延胡索对照药材0.2-2g,加体积浓度90-100%的甲醇溶液30-80ml,超声处理15-60分钟,滤过,滤液蒸干,残渣加水5-20ml使溶解,加浓氨试液调至碱性,用乙醚振摇提取1-5次,每次5-20ml,合并乙醚液,蒸干,残渣加体积浓度90-100%的甲醇1-3ml使溶解,即得对照药材溶液,另取延胡索乙素对照品,加体积浓度90-100%甲醇溶液制成每1ml含0.1-1mg胡索乙素对照品的溶液,即得对照品溶液;
鉴别:照薄层色谱法试验,分别精密吸取上述对照品药材溶液、对照品溶液和供试品溶液各1-10μ1,分别点于同一硅胶g薄层板上,以浓氨水饱和的体积比为5-15︰1-5的甲苯-丙酮为展开剂,展开,然后取出,晾干,置碘缸中1-5分钟后取出,挥尽板上吸附的碘后,置紫外光灯(365nm)下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点;或
d、秦艽的鉴别:
供试品溶液的制备:取所述中药组合物0.5-4g,加体积浓度90-100%甲醇溶液5-30ml,超声处理10-60分钟,放冷,滤过,取续滤液,即得供试品溶液;
对照品溶液的制备:另取龙胆苦苷对照品,加体积浓度90-100%甲醇制成每1ml含0.2-2mg龙胆苦苷对照品的溶液,即得对照品溶液;
鉴别:照薄层色谱法试验,吸取上述供试品溶液和对照品溶液各1-10μl,分别点于同一硅胶gf254薄层板上,以体积比为5-20︰1-5︰0.5-2的乙酸乙酯-甲醇-水溶液为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视;供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点;或
e、秦艽的鉴别:
供试品溶液的制备:取所述中药组合物0.5-4g,加体积浓度90-100%甲醇溶液5-30ml,超声处理10-60分钟,放冷,滤过,取续滤液,即得供试品溶液;
对照品溶液的制备:另取栎瘿酸对照品,加体积浓度90-100%三氯甲烷溶液制成每1ml含0.2-2mg的溶液,即得对照品溶液;
鉴别:照薄层色谱法试验,吸取上述供试品溶液5-20μ1和对照品溶液1-5μ1,分别点于同一硅胶g薄层板上,以体积比为40-60︰0.5-2︰0.2-1的二氯甲烷-甲醇-甲酸为展开剂,展开,取出,晾干,喷以8-12vt%硫酸乙醇溶液,在102-108℃下加热至斑点显色清晰;供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点;或
f、羌活的鉴别:
供试品溶液的制备:取所述中药组合物0.5-4g,加体积浓度90-100%甲醇溶液5-30ml,超声处理10-60分钟,放冷,滤过,取续滤液,即得供试品溶液;
对照品溶液的制备:另取紫花前胡苷对照品,加体积浓度90-100%甲醇溶液制成每1ml含0.2-2mg紫花前胡苷对照品的溶液,即得对照品溶液;
鉴别:照薄层色谱法试验,吸取上述供试品溶液和对照品溶液各1-10μ1,分别点于同一硅胶g薄层板上,以体积比为5-10:1-3的二氯甲烷-甲醇为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视;供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。
优选地,所述中药组合物包括如下重量份的原料药组成:骨碎补10份、狗脊6份、杜仲6份、桂枝6份、鹿角4份、延胡索7份、续断7份、秦艽7份、青风藤9份和羌活6份;或
骨碎补8份、狗脊4份、杜仲4份、桂枝4份、鹿角2份、延胡索5份、续断5份、秦艽5份、青风藤7份和羌活4份;或
骨碎补12份、狗脊8份、杜仲8份、桂枝8份、鹿角6份、延胡索9份、续断9份、秦艽9份、青风藤11份和羌活8份。
进一步地,所述药物组合物添加常规辅料按照常规工艺直接或间接制成临床上可接受的片剂、颗粒剂、胶囊剂、口服液或注射剂。
优选地,所述中药组合物的制备方法,包括如下步骤:
(1)按照选定重量份数称取桂枝和羌活,混合,加适量水,水蒸汽蒸馏,分别收集挥发油和水提取液备用,取挥发油,加β-环糊精和水,包合,干燥,得包合物,备用;
(2)按照选定重量份数称取狗脊、杜仲、鹿角和续断,混合,加水提取,得提取液,并与步骤(1)制得的提取液合并,减压浓缩,得清膏,备用;
(3)按照选定重量份数称取骨碎补、青风藤、秦艽和延胡索,用20-95vt%乙醇溶液提取,浓缩后与上述清膏合并;继续浓缩,干燥,粉碎成细粉,加入上述包合物,混匀,即得。
进一步地,步骤(1)包合过程中,挥发油、β-环糊精与水的用量比为:1ml:4-10g:40-100ml;
包合时间为20-60分钟,包合后将包合物冷藏12-48小时。
优选地,步骤(1)水蒸气蒸馏过程中,加入8-12倍量的水,水蒸气蒸馏4-8小时;
步骤(2)水提取过程中,加水煎煮1-3次,每次1-2小时,每次加4-10倍量的水;
步骤(3)乙醇提取过程中,用体积浓度60%-80%乙醇溶液加热回流提取1-3次,每次1-2小时,每次加体积浓度60%-80%乙醇溶液6-12倍量,提取液滤过,合并。
本发明还提供了所述的质量检测方法在中药组合物或中药制剂质量检测中的应用。
本发明技术方案,具有如下优点:
1.本发明所述的补肾舒脊的中药组合物的质量检测方法,通过测定所述中药组合的有效成分柚皮苷的含量,实现了对产品质量的有效监控,在柚皮苷含量测试方法的供试品溶液制备中,结合该中药组合物中药物成分复杂和多样的特点,通过多次筛选得到合适的提取方法,先加入甲醇溶液,超声提取,过滤,滤液蒸干,加水溶解,柱层析提纯,收集洗脱液蒸干,加甲醇溶液溶解定容,得供试品溶液,在此基础上,筛选得到合适的流动相组成,利用hplc正相色谱法或反相色谱法,选择合适的色谱条件,最终能够将供试品溶液中的柚皮苷与其他杂质峰有效地分离开来,有效地克服了因中药药味繁多、各类成分复杂且相互干扰而不能全面的、清晰地对该中药组合物的质量进行检测的缺陷,得到该中药组合物中的有效成分柚皮苷的色谱峰,确定的hplc含量测定方法不仅专属性强,线性关系、精密度、回收率均满足药物分析要求,从而实现了通过测定有效成分柚皮苷的含量来控制中药组合物或中药制剂的含量的目的,能够全面、清楚地对所述中药组合物进行质量检测,进一步实现了对所述中药组合物质量的控制,且该质量检测方法具有简单快速、稳定可靠、精密度高、易于掌握的优势。
2.本发明所述的补肾舒脊的中药组合物的质量检测方法,通过测定青藤碱的含量,提高了对所述中药组合物进行质量监控的全面性和可靠性;通过筛选得到合适的提取方法对该中药组合物进行提取制得供试品溶液,选择合适的流动相,最终确定的hplc含量测定方法不仅专属性强,线性关系、精密度、回收率均满足药物分析要求,能够快速、准确的获知该产品的质量情况,更可以有效的对产品的质量性能进行监控。
3.本发明所述的补肾舒脊的中药组合物的质量检测方法,通过tlc法对延胡索、秦艽或羌活进行鉴别,使得对所述补肾舒脊的中药组合物的质量检测更加全面、清楚,更完善地表征了药物的有效活性组分及其质量,有利于对活性成分的全面监控,进一步保证了所述补肾舒脊的中药组合物质量的稳定性、一致性和可控性,并确保了所述中药组合物的安全性和有效性。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为本发明实施例4中所述中药组合物的薄层色谱图;图中从左到右依次为:延胡索阴性样品、供试品1、供试品2、供试品3、延胡索乙素对照品、延胡索对照药材;
图2为本发明实施例5中所述中药组合物的薄层色谱图;图中从左到右依次为:秦艽阴性样品、供试品1、供试品2、供试品3、龙胆苦苷对照品;
图3为本发明实施例6中所述中药组合物的薄层色谱图;图中从左到右依次为:秦艽阴性样品、供试品1、供试品2、供试品3、栎瘿酸对照品;
图4为本发明实施例7中所述中药组合物的薄层色谱图;图中从左到右依次为:羌活阴性样品、供试品1、供试品2、供试品3、紫花前胡苷对照品;
图5是实验例1中试验组1和试验组2以及对比组1的含量测定高效液相色谱图;a为试验组1的供试品;b为试验组2供试品;c为对比组1的供试品;
图6是实验例1中对比组2-4的含量测定高效液相色谱图;a为对比组2的供试品;b为对比组2的阴性对照品溶液色谱图;c为对比组3的供试品溶液色谱图;d为对比组4的供试品溶液色谱图;
图7是实验例2中的高效液相色谱图;其中a为对照品溶液色谱图;b为供试品溶液色谱图,c为阴性对照品溶液色谱图;
图8是实验例3中的高效液相色谱图;其中a为对照品溶液色谱图;b为供试品溶液色谱图,c为阴性对照品溶液色谱图;
图9是实验例4中的高效液相色谱图;其中a为对照品溶液色谱图;b为供试品溶液色谱图,c为阴性对照品溶液色谱图;
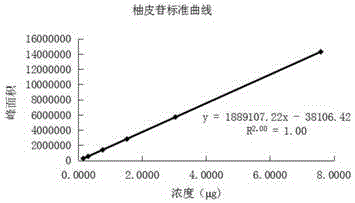
图10是实验例2线性关系的考察中得到的柚皮苷标准曲线图;
图11是实验例3线性关系的考察中得到的柚皮苷线性曲线图;
图12是实验例4线性关系的考察中得到的青藤碱线性曲线图。
具体实施方式
实施例1颗粒剂
处方:骨碎补10g、狗脊6g、杜仲6g、桂枝6g、鹿角4g、延胡索7g、续断7g、秦艽7g、青风藤9g和羌活6g;
制法:按照选定重量份数称取骨碎补、狗脊、杜仲、桂枝、鹿角、延胡索、续断、秦艽、青风藤和羌活,备用;取桂枝、羌活两味,加10倍量水,水蒸汽蒸馏6小时,提取挥发油,收集挥发油,水提取液过滤,备用;按照挥发油、β-环糊精和水为1ml:6g:60ml,向挥发油中加入β-环糊精和水,搅拌,包合30分钟,冷藏24小时,抽滤,40℃干燥,得挥发油包合物,备用;取狗脊、杜仲、鹿角、续断四味,加水煎煮2次,每次2小时,每次加6倍量水,煎液滤过,与上述提取液合并,减压浓缩至相对密度为1.15(50℃)的清膏,备用;取骨碎补、青风藤、秦艽、延胡索四味,用体积浓度70%乙醇加热回流提取3次,每次2小时,每次加体积浓度70%乙醇8倍量,提取液滤过,合并,回收乙醇,浓缩至相对密度为1.15(50℃),与上述清膏合并;继续浓缩至相对密度为1.30(50℃)的清膏,干燥,粉碎成细粉;取上述细粉,加入挥发油包合物、适量糊精及适量阿斯帕坦,混匀,制成颗粒1000g,即得。
a、柚皮苷的含量测定
供试品溶液的制备:取上述制得的颗粒剂,约1.5g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,超声处理(功率250w,频率40khz)0.5小时,取出,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取滤液10ml,蒸干,残渣加水5ml,溶解,放冷,过聚酰胺色谱柱(60~80目,2g,内径1.8cm),以25ml水液洗脱,弃去水液,再用甲醇25ml洗脱,收集洗脱液蒸干,再用甲醇定容至5ml,摇匀,滤过,即得;
对照品溶液的制备:取柚皮苷对照品适量,精密称定,加甲醇溶液制成每1ml含75μg柚皮苷对照品的溶液,即得;
色谱条件与系统适用性试验:照高效液相色谱法测定,以氨基键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,按照流动相a:流动相b的体积比为90%:10%;检测波长为283nm,理论塔板数按柚皮苷计算应不低于2000;
测定法:分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定并计算;
本品每日服用剂量含骨碎补以柚皮苷(c27h32o14)计,不得少于17.64mg。
实施例2颗粒剂
处方:骨碎补8g、狗脊4g、杜仲4g、桂枝4g、鹿角2g、延胡索5g、续断5g、秦艽5g、青风藤7g和羌活4g;
制法:按照选定重量份数称取骨碎补、狗脊、杜仲、桂枝、鹿角、延胡索、续断、秦艽、青风藤和羌活,备用;取桂枝、羌活两味,加12倍量水,水蒸汽蒸馏4小时,提取挥发油,收集挥发油,水提取液过滤,备用;按照挥发油、β-环糊精和水为1ml:4g:80ml,向挥发油中加入β-环糊精和水,包合60分钟,冷藏12小时,抽滤,50℃干燥,得挥发油包合物,备用;取狗脊、杜仲、鹿角、续断四味,加水煎煮1次,每次2小时,每次加4倍量水,煎液滤过,与上述提取液合并,减压浓缩至相对密度为1.15(50℃)的清膏,备用;取骨碎补、青风藤、秦艽、延胡索四味,用体积浓度70%乙醇加热回流提取3次,每次2小时,每次加体积浓度60%乙醇8倍量,提取液滤过,合并,回收乙醇,浓缩至相对密度为1.15(50℃),与上述清膏合并;继续浓缩至相对密度为1.30(50℃)的清膏,干燥,粉碎成细粉;取上述细粉,加入挥发油包合物、适量糊精及适量阿斯帕坦,混匀,制成颗粒1000g,即得。
a、柚皮苷的含量测定
供试品溶液的制备:取上述制得的颗粒剂,约1.5g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,超声处理(功率250w,频率40khz)0.5小时,取出,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取滤液10ml,蒸干,残渣加水5ml,溶解,放冷,过聚酰胺色谱柱(60~80目,2g,内径1.8cm),以25ml水液洗脱,弃去水液,再用甲醇25ml洗脱,收集洗脱液蒸干,再用甲醇定容至5ml,摇匀,滤过,即得;
对照品溶液的制备:取柚皮苷对照品适量,精密称定,加甲醇溶液制成每1ml含75μg柚皮苷对照品的溶液,即得;
色谱条件与系统适用性试验:照高效液相色谱法测定,以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以0.1%磷酸溶液为流动相b,按照流动相a:流动相b的体积比为20%:80%;检测波长为283nm,理论塔板数按柚皮苷计算应不低于2000;
测定法:分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定并计算;
本品每日服用剂量含骨碎补以柚皮苷(c27h32o14)计,不得少于17.64mg。
实施例3颗粒剂
组成:骨碎补12g、狗脊8g、杜仲8g、桂枝8g、鹿角6g、延胡索9g、续断9g、秦艽9g、青风藤11g和羌活8g;
制法:按照选定重量份数称取骨碎补、狗脊、杜仲、桂枝、鹿角、延胡索、续断、秦艽、青风藤和羌活,备用;取桂枝、羌活两味,加8倍量水,水蒸汽蒸馏8小时,提取挥发油,收集挥发油,水提取液过滤,备用;按照挥发油、β-环糊精和水为1ml:10g:40ml,向挥发油中加入β-环糊精和水,包合20分钟,冷藏48小时,抽滤,30℃干燥,得挥发油包合物,备用;取狗脊、杜仲、鹿角、续断四味,加水煎煮3次,每次1小时,每次加10倍量水,煎液滤过,与上述提取液合并,减压浓缩至相对密度为1.10(50℃)的清膏,备用;取骨碎补、青风藤、秦艽、延胡索四味,用体积浓度80%乙醇加热回流提取1次,每次1小时,每次加体积浓度60%乙醇12倍量,提取液滤过,合并,回收乙醇,浓缩至相对密度为1.05(50℃),与上述清膏合并;继续浓缩至相对密度为1.20(50℃)的清膏,干燥,粉碎成细粉;取上述细粉,加入上述挥发油包合物、适量糊精及适量阿斯帕坦,混匀,制成颗粒1000g,即得。
b、青藤碱的含量测定
供试品溶液的制备:取上述制得的颗粒剂,约1g,精密称定,置具塞锥形瓶中,精密加入体积浓度70%乙醇10ml,密塞,称定重量,超声处理(功率250w,频率40khz)20分钟,放冷,再称定重量,加体积浓度70%乙醇补足减失的重量,摇匀,滤过,取续滤液,即得;
对照品溶液的制备:取青藤碱对照品适量,精密称定,加甲醇制成每1ml含0.2mg青藤碱对照品的溶液,即得;
色谱条件与系统适用性试验:照高效液相色谱法测定,以十八烷基硅烷键合硅胶为填充剂;以甲醇为流动相a,以磷酸盐缓冲液(0.005mol/l磷酸氢二钠溶液,以0.005mol/l的磷酸二氢钠调节ph值至8.0,再以1%三乙胺调节ph值至9.0)为流动相b,按照流动相a:流动相b的体积比为32%:68%;检测波长为262nm,理论塔板数按青藤碱计算应不低于1500;
测定法:分别精密吸取对照品溶液与供试品溶液各5μl,注入液相色谱仪,测定并计算;
本品每日服用剂量含青风藤以青藤碱(c27h32o14)计,不得少于18.9mg。
实施例4片剂
组成:骨碎补12g、狗脊8g、杜仲8g、桂枝8g、鹿角2g、延胡索5g、续断5g、秦艽9g、青风藤11g和羌活8g;
制法:按照选定重量份数称取骨碎补、狗脊、杜仲、桂枝、鹿角、延胡索、续断、秦艽、青风藤和羌活,备用;取桂枝、羌活两味,加8倍量水,水蒸汽蒸馏8小时,提取挥发油,收集挥发油,水提取液过滤,备用;按照挥发油、β-环糊精和水为1ml:10g:40ml,向挥发油中加入β-环糊精和水,包合20分钟,冷藏48小时,抽滤,30℃干燥,得挥发油包合物,备用;取狗脊、杜仲、鹿角、续断四味,加水煎煮3次,每次1小时,每次加10倍量水,煎液滤过,与上述提取液合并,减压浓缩至相对密度为1.10(50℃)的清膏,备用;取骨碎补、青风藤、秦艽、延胡索四味,用体积浓度80%乙醇加热回流提取1次,每次1小时,每次加体积浓度60%乙醇12倍量,提取液滤过,合并,回收乙醇,浓缩至相对密度为1.05(50℃),与上述清膏合并;继续浓缩至相对密度为1.20(50℃)的清膏,干燥,粉碎成细粉;取上述细粉,加入上述挥发油包合物、适量糊精及适量阿斯帕坦,混匀,压制成片剂,即得。
c、延胡索的鉴定
供试品溶液的制备:供试品溶液的制备:取上述制得的片剂2g,加甲醇溶液50ml,超声处理30分钟,滤过,滤液蒸干,残渣加水10ml使溶解,加浓氨试液调至碱性,用乙醚振摇提取3次,每次10ml,合并乙醚液,蒸干,残渣加甲醇1ml使溶解,即得;
对照品药材溶液和对照品溶液的制备:取延胡索对照药材1g,加甲醇溶液50ml,超声处理30分钟,滤过,滤液蒸干,残渣加水10ml使溶解,加浓氨试液调至碱性,用乙醚振摇提取3次,每次10ml,合并乙醚液,蒸干,残渣加甲醇1ml使溶解,即得延胡索对照药材溶液。另取延胡索乙素对照品,加甲醇制成每1ml含0.5mg延胡索乙素的溶液,即得延胡索乙素对照品溶液;
延胡索阴性样品溶液:按照供试品溶液的制备方法制备,其区别仅在于所述处方中不含延胡索;
鉴别:照薄层色谱法试验,分别精密吸取上述对照品溶液、供试品溶液和延胡索阴性样品溶液各3μ1,分别点于同一硅胶g薄层板上,以浓氨水饱和的体积比为9:2的甲苯-丙酮为展开剂,展开,然后取出,晾干,置碘缸中约3分钟后取出,挥尽板上吸附的碘后,置紫外光灯(365nm)下检视;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点;经延胡索阴性样品对照,阴性样品无干扰,所述谱图见图1所示,其中1为延胡索阴性样品,2、3、4为供试品样品,5为延胡索乙素对照品,6为延胡索对照药材。
实施例5胶囊剂
组成:骨碎补8g、狗脊8g、杜仲8g、桂枝8g、鹿角2g、延胡索5g、续断5g、秦艽5g、青风藤7g和羌活4g;
制法:按照选定重量份数称取骨碎补、狗脊、杜仲、桂枝、鹿角、延胡索、续断、秦艽、青风藤和羌活,备用;取桂枝、羌活两味,加12倍量水,水蒸汽蒸馏4小时,提取挥发油,收集挥发油,水提取液过滤,备用;按照挥发油、β-环糊精和水为1ml:8g:80ml,向挥发油中加入β-环糊精和水,包合60分钟,冷藏12小时,抽滤,50℃干燥,得挥发油包合物,备用;取狗脊、杜仲、鹿角、续断四味,加水煎煮1次,每次2小时,每次加4倍量水,煎液滤过,与上述提取液合并,减压浓缩至相对密度为1.15(50℃)的清膏,备用;取骨碎补、青风藤、秦艽、延胡索四味,用体积浓度70%乙醇加热回流提取3次,每次2小时,每次加体积浓度60%乙醇8倍量,提取液滤过,合并,回收乙醇,浓缩至相对密度为1.15(50℃),与上述清膏合并;继续浓缩至相对密度为1.30(50℃)的清膏,干燥,粉碎成细粉;取上述细粉,加入上述挥发油包合物、适量糊精及适量阿斯帕坦,混匀,制成颗粒,装入胶囊,即得。
d、秦艽的鉴别
供试品溶液的制备:取所述胶囊剂2g,去胶囊壳,取颗粒,加甲醇溶液10ml,超声处理15分钟,放冷,滤过,取续滤液,即得供试品溶液;
对照品溶液的制备:取龙胆苦苷对照品,加甲醇制成每1ml含1mg龙胆苦苷对照品的溶液,即得对照品溶液;
秦艽阴性样品溶液:按照供试品溶液的制备方法制备,其区别仅在于所述中药组合物中不含有秦艽;
鉴别:照薄层色谱法试验,吸取上述供试品溶液和对照品溶液各3μl,分别点于同一硅胶gf254薄层板上,以体积比为10︰2︰1的乙酸乙酯-甲醇-水溶液为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视;供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点;经秦艽阴性样品对照,阴性样品无干扰,所述谱图见图2所示,其中1为秦艽阴性样品,2、3、4为供试品,5为龙胆苦苷对照品。
实施例6中药组合物
处方:骨碎补10g、狗脊6g、杜仲6g、桂枝6g、鹿角4g、延胡索7g、续断7g、秦艽7g、青风藤9g和羌活6g;
制法:按照选定重量份数称取骨碎补、狗脊、杜仲、桂枝、鹿角、延胡索、续断、秦艽、青风藤和羌活,备用;取桂枝、羌活两味,加10倍量水,水蒸汽蒸馏6小时,提取挥发油,收集挥发油,水提取液过滤,备用;按照挥发油、β-环糊精和水为1ml:4g:80ml,向挥发油中加入β-环糊精和水,包合30分钟,冷藏24小时,抽滤,40℃干燥,备用;取狗脊、杜仲、鹿角、续断四味,加水煎煮2次,每次2小时,每次加6倍量水,煎液滤过,与上述提取液合并,减压浓缩至相对密度为1.15(50℃)的清膏,得挥发油包合物,备用;取骨碎补、青风藤、秦艽、延胡索四味,用体积浓度70%乙醇加热回流提取3次,每次2小时,每次加体积浓度70%乙醇8倍量,提取液滤过,合并,回收乙醇,浓缩至相对密度为1.15(50℃),与上述清膏合并;继续浓缩至相对密度为1.30(50℃)的清膏,干燥,粉碎成细粉;取上述细粉,加入上述挥发油包合物混匀,制得中药组合物。
e、秦艽的鉴别
供试品溶液的制备:取所述中药组合物2g,加甲醇溶液10ml,超声处理15分钟,放冷,滤过,取续滤液,即得供试品溶液;
对照品溶液的制备:另取栎瘿酸对照品,加三氯甲烷溶液制成每1ml含0.5mg栎瘿酸对照品的溶液,即得对照品溶液;
秦艽阴性样品溶液:按照供试品溶液的制备方法制备,其区别仅在于处方中不含有秦艽;
鉴别:照薄层色谱法试验,吸取上述供试品溶液10μ1和对照品溶液1μ1,分别点于同一硅胶g薄层板上,以体积比为50:1:0.5的二氯甲烷-甲醇-甲酸为展开剂,展开,取出,晾干,喷以10vt%硫酸乙醇溶液,在105℃下加热至斑点显色清晰;供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点;经阴性样品对照,阴性样品无干扰,所述谱图见图3所示,其中1为秦艽阴性样品,2、3、4为供试品,5为栎瘿酸对照品。
实施例7
处方:骨碎补10g、狗脊4g、杜仲4g、桂枝4g、鹿角4g、延胡索7g、续断7g、秦艽7g、青风藤11g和羌活8g;
制法:按照选定重量份数称取骨碎补、狗脊、杜仲、桂枝、鹿角、延胡索、续断、秦艽、青风藤和羌活,备用;取桂枝、羌活两味,加10倍量水,水蒸汽蒸馏6小时,提取挥发油,收集挥发油,水提取液过滤,备用;按照挥发油、β-环糊精和水为1ml:6g:60ml,向挥发油中加入β-环糊精和水,包合30分钟,冷藏24小时,抽滤,40℃干燥,得挥发油包合物,备用;取狗脊、杜仲、鹿角、续断四味,加水煎煮2次,每次2小时,每次加6倍量水,煎液滤过,与上述提取液合并,减压浓缩至相对密度为1.10(50℃)的清膏,备用;取骨碎补、青风藤、秦艽、延胡索四味,用体积浓度70%乙醇加热回流提取3次,每次2小时,每次加体积浓度70%乙醇8倍量,提取液滤过,合并,回收乙醇,浓缩至相对密度为1.10(50℃),与上述清膏合并;继续浓缩至相对密度为1.30(50℃)的清膏,干燥,粉碎成细粉;与上述挥发油包合物混匀,制得中药组合物。
f、羌活的鉴别
供试品溶液的制备:取所述中药组合物2g,加甲醇溶液10ml,超声处理15分钟,放冷,滤过,取续滤液,即得供试品溶液;
对照品溶液的制备:另取紫花前胡苷对照品,加甲醇溶液制成每1ml含0.5mg紫花前胡苷对照品的溶液,即得对照品溶液;
阴性样品溶液:按照供试品溶液的制备方法制备,其区别仅在于所述中药组合物处方中不含有羌活;
鉴别:照薄层色谱法试验,吸取上述供试品溶液和对照品溶液各2μ1,分别点于同一硅胶g薄层板上,以体积比为8︰2的二氯甲烷-甲醇为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视;供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点;经阴性样品对照,阴性样品无干扰,所述谱图见图4所示,其中1为羌活阴性样品,2、3、4为供试品,5为紫花前胡苷对照品。
实验例1柚皮苷含量测定方法的考察
取实施例1制得的颗粒剂,平行分为6组,分别为试验组1,试验组2,对比组1,对比组2、对比组3和对比组4,分别按照如下方法进行柚皮苷含量测定。
(1)试验组1:取上述颗粒剂按照实施例1中“供试品溶液的制备”方法和“对照品溶液的制备”方法,制得供试品溶液和对照品溶液,按实施例1中记载的“处方”和“制法”制备颗粒剂,不同之处在于不加骨碎补,即得骨碎补阴性对照品,再按实施例1记载的“供试品溶液的制备”方法制备骨碎补阴性对照品溶液,然后按照实施例1公开的“色谱条件”和“测定法”进行图谱测定,结果如图5-a所示,供试品溶液中柚皮苷峰与其他杂质峰的分离度均大于1.5,分离度满足要求。
(2)试验组2:取上述颗粒剂按照实施例2中“供试品溶液的制备”方法和“对照品溶液的制备”方法,制得供试品溶液和对照品溶液,按实施例2中记载的“处方”和“制法”制备颗粒剂,不同之处在于不加骨碎补,即得骨碎补阴性对照品,再按实施例2记载的“供试品溶液的制备”方法制备骨碎补阴性对照品溶液,然后按照实施例2公开的“色谱条件”和“测定法”进行图谱测定,结果如图5-b所示,供试品溶液中柚皮苷峰与其他杂质峰的分离度均大于1.5,分离度满足要求。
(3)对比组1(药典法)
供试品溶液的制备:取上述制得的颗粒剂,约1.5g,精密称定,置具塞锥形瓶中,精密加入甲醇180ml,加热回流3h,取出,放冷,滤过,取滤液置于200ml量瓶中,用甲醇分多次洗涤容器,洗涤液入同一量瓶中,加甲醇至刻度,摇匀,即得;
对照品溶液的制备:同实施例2对照品溶液的制备方法。
骨碎补阴性对照品,按实施例2中记载的“处方”和“制法”制备颗粒剂,不同之处在于不加骨碎补,即得骨碎补阴性对照品,再按本对比组中记载的供试品溶液制备方法制备骨碎补阴性对照品溶液。
色谱条件与系统适用性试验:照高效液相色谱法测定,以十八烷基硅烷键合硅胶为填充剂;以甲醇-醋酸-水(35:4:65)为流动相;检测波长为283nm,理论塔板数按柚皮苷计算应不低于2000;
测定法:分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定并计算,如图5-c所示,供试品溶液中柚皮苷峰有肩峰,未完全分开,分离度明显不合格。
(4)对比组2:取上述颗粒剂按照实施例2中“供试品溶液的制备”方法和“对照品溶液的制备”方法,制得供试品溶液和对照品溶液,按实施例2中记载的“处方”和“制法”制备颗粒剂,不同之处在于不加骨碎补,即得骨碎补阴性对照品,再按实施例2记载的供试品溶液制备方法制备骨碎补阴性对照品溶液,然后按照如下色谱条件进行图谱测定。
色谱条件:照高效液相色谱法测定,以十八烷基硅烷键合硅胶为填充剂;以甲醇-水(32:68)为流动相;检测波长为283nm,理论塔板数按柚皮苷计算应不低于2000。
如图6-a和6-b所示,分别为供试品溶液和骨碎补阴性对照品溶液的色谱图,骨碎补阴性对照品在柚皮苷对应的保留时间(29min)处干扰明显。
(5)对比组3:取上述颗粒剂按照实施例2中“供试品溶液的制备”方法和“对照品溶液的制备”方法,制得供试品溶液和对照品溶液,按实施例2中记载的“处方”和“制法”制备颗粒剂,不同之处在于不加骨碎补,即得骨碎补阴性对照品,再按实施例2记载的供试品溶液制备方法制备骨碎补阴性对照品溶液,然后按照如下色谱条件进行图谱测定。
色谱条件:照高效液相色谱法测定,以十八烷基硅烷键合硅胶为填充剂;以乙腈-水(15:85)为流动相;检测波长为283nm,理论塔板数按柚皮苷计算应不低于2000。
如图6-c所示,供试品溶液中柚皮苷峰有肩峰,未完全分开,分离度明显不合格。
(6)对比组4取上述颗粒剂按照实施例2中“供试品溶液的制备”方法和“对照品溶液的制备”方法,制得供试品溶液和对照品溶液,按实施例2中记载的“处方”和“制法”制备颗粒剂,不同之处在于不加骨碎补,即得骨碎补阴性对照品,再按实施例2记载的供试品溶液制备方法制备骨碎补阴性对照品溶液,然后按照如下色谱条件进行图谱测定。
色谱条件:照高效液相色谱法测定,以十八烷基硅烷键合硅胶为填充剂;以乙腈-含0.1vt%磷酸的水溶液(17:83)为流动相;检测波长为283nm,理论塔板数按柚皮苷计算应不低于2000。
如图6-d所示,此图为供试品溶液图谱的放大图,供试品溶液中柚皮苷峰有干扰,分离度明显不合格。
(7)结果分析:试验组1和试验组2以及对比组1-4的色谱分析结果显示,由于该中药组合物中药药味繁多、成分复杂且相互干扰,使用对比组1-4中的方法进行检测时,柚皮苷峰与其他杂质峰的分离度均小于1.5,分离效果差,不能满足色谱分析要求,而试验组1和试验组2中,柚皮苷峰与其他杂质峰的分离度均大于1.5,其中试验组1的分离度为9.964,试验组2的分离度为2.718,满足色谱分析要求,且与试验组2相比,试验组1的正相色谱分析中柚皮苷峰与其他杂质峰的分离度明显提高,且试验组1的耐用性较高,具有明显提高的分离效果。
实验例2柚皮苷正相色谱法含量测定的方法学考察
1专属性考察
①按照实施例1记载的处方和制法制备颗粒剂,然后按照实施例1中供试品溶液的制备方法和对照品溶液的制备方法分别制得供试品溶液和对照品溶液;②按实施例1中记载的处方比例和制法,制备不含骨碎补的颗粒剂,即骨碎补阴性对照品,再按实施例1记载的供试品溶液制备方法制备骨碎补阴性对照品溶液。
按实施例1中的色谱条件,分别取供试品溶液、对照品溶液和骨碎补阴性对照品溶液进样色谱柱,结果见图7所示,供试品溶液中柚皮苷峰与其他杂质峰的分离度均大于1.5,说明分离情况较好,且骨碎补阴性对照品溶液在与柚皮苷相同保留时间处均未显色谱峰,故认为无干扰,专属性强。
2线性关系的考察
精密吸取上述对照品溶液,稀释得一系列标准品溶液,精密吸取10μl标准品溶液分别注入液相色谱仪,按上述色谱条件测定峰面积,以峰面积积分值为纵坐标,对照品的进样量为横坐标,绘制柚皮苷的标准曲线,如图10所示,回归方程:y=1889107.22x-38106.42,r2=1.00;结果表明,实施例1所述的检测方法可有效将柚皮苷进行检测,柚皮苷0.1518μg-7.5890μg范围内进样量与峰面积呈良好线性关系。
3精密度试验
精密量取上述对照品溶液,按实施例1中的色谱条件重复进样6次,测定柚皮苷的峰面积,计算峰面积的rsd值。其中,柚皮苷峰面积的rsd值为0.89%。结果表明,本色谱条件下仪器精密度良好。
4重复性试验
取上述颗粒剂,按实施例1中记载的方法平行操作,制备供试品溶液六份,并测定柚皮苷的峰面积,计算含量及rsd值,其中,柚皮苷含量的rsd值为1.25%。结果表明,本方法重复性良好。
5回收率试验
采用100%加样回收法。精密称取上述颗粒剂约1.5g,共6份,按实施例1供试品溶液的制备方法制得六份供试品溶液,分别加入一定量的对照品溶液,按照实施例1记载的色谱条件进行测定,按下式计算回收率,回收率=(实测值-供试品含量×取样量)/加入量×100%,其中,柚皮苷的平均回收率为100.89%,rsd值为1.44%。结果表明,本方法的回收率符合要求。
实验例3柚皮苷反相色谱法含量测定的方法学考察
1专属性考察
①按照实施例2记载的处方和制法制备颗粒剂,然后按照实施例2中供试品溶液的制备方法和对照品溶液的制备方法分别制得供试品溶液和对照品溶液;②按实施例2中记载的处方比例和制法,制备不含骨碎补的颗粒剂,即骨碎补阴性对照品,再按实施例2记载的供试品溶液制备方法制备骨碎补阴性对照品溶液。
按实施例2中的色谱条件,分别取供试品溶液、对照品溶液和骨碎补阴性对照品溶液进样色谱柱,结果见图8所示,供试品溶液中柚皮苷峰与其他杂质峰的分离度均大于1.5,说明分离情况较好,且骨碎补阴性对照品溶液在与柚皮苷相同保留时间处均未显色谱峰,故认为无干扰,专属性强。
2线性关系的考察
精密吸取上述对照品溶液,稀释得一系列标准品溶液,精密吸取10μl分别注入液相色谱仪,按上述色谱条件测定峰面积,以峰面积积分值为纵坐标,对照品的进样量为横坐标,绘制柚皮苷的标准曲线,如图11所示,回归方程:y=1651822.5190x+154941.2281,r2=0.9998;结果表明,实施例2所述的检测方法可有效将柚皮苷进行检测,柚皮苷0.261μg-20.88μg范围内进样量与峰面积呈良好线性关系。
3精密度试验
精密量取上述对照品溶液,按实施例2中的色谱条件重复进样6次,测定柚皮苷的峰面积,计算峰面积的rsd值。其中,柚皮苷峰面积的rsd值为0.49%。结果表明,本色谱条件下仪器精密度良好。
4重复性试验
取上述颗粒剂,按实施例2中记载的方法平行操作,制备供试品溶液六份,并测定人柚皮苷的峰面积,计算含量及rsd值,其中,柚皮苷含量的rsd值为0.85%。结果表明,本方法重复性良好。
5回收率试验
采用100%加样回收法。精密称取上述颗粒剂约1.5g,共6份,按实施例2供试品溶液的制备方法制得六份供试品溶液,分别加入一定量的对照品溶液,按照实施例2记载的色谱条件进行测定,按下式计算回收率,回收率=(实测值-供试品含量×取样量)/加入量×100%,其中,柚皮苷的平均回收率为98.8%,rsd值为1.80%。结果表明,本方法的回收率符合要求。
实验例4青藤碱含量测定的方法学考察
1专属性考察
①按照实施例3记载的处方和制法制备颗粒剂,然后按照实施例3中供试品溶液的制备方法和对照品溶液的制备方法分别制得供试品溶液和对照品溶液;②按实施例3中记载的处方比例和制法,分别制备不含青风藤的颗粒剂,即青风藤阴性对照品,再按实施例3记载的供试品溶液制备方法分别制备青风藤阴性对照品溶液。
按实施例3中的色谱条件,分别取供试品溶液、对照品溶液、青风藤阴性对照品溶液进样色谱柱,结果见图9所示,供试品溶液中青藤碱与其他杂质峰的分离度均大于1.5,说明分离情况较好,青风藤阴性对照品溶液在与青藤碱相同保留时间处未显色谱峰,故认为无干扰,专属性强。
2线性关系考察
精密吸取青藤碱对照品溶液稀释得一系列标准品溶液,取标准品溶液10μl,分别注入液相色谱仪,按上述色谱条件测定峰面积,以峰面积积分值为纵坐标,青藤碱的量为横坐标,绘制标准曲线,如图12所示,结果表明,青藤碱在0.108μg-15.05μg范围内呈良好线性关系,得回归方程为y=995781.4686x+43146.1334,r2=0.9998,可用外标一点法进行计算。
3精密度试验
精密量取上述对照品溶液,按实施例3中的色谱条件重复进样6次,并测定青藤碱峰面积,计算峰面积的rsd值。rsd值为0.74%。结果表明,本色谱条件下仪器精密度良好。
4重复性试验
取上述颗粒剂,按实施例3中记载的方法平行操作,制备供试品溶液六份,并测定青藤碱的峰面积,计算含量及rsd值,其中,青藤碱含量的rsd值为0.24%。结果表明,本方法重复性良好。
5回收率试验
采用100%加样回收法。精密称取上述颗粒剂约1g,共6份,按实施例3供试品溶液的制备方法制得六份供试品溶液,分别精密加入一定量的对照品溶液,按照实施例3记载的色谱条件进行测定,按下式计算回收率,回收率=(实测值-供试品含量×加入量)/加入量×100%,6份样品的平均回收率为99.9%,rsd%为1.08%。结果表明,本方法回收率符合要求。
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!