经鼻给药用医药组合物的制作方法
本发明涉及一种医药组合物,其为经鼻给药用医药组合物,治疗效果高且副作用得到抑制,从而对痴呆症的预防或治疗有用。
背景技术:
痴呆症有因脑血管障碍而发病的脑血管性痴呆症、和异常蛋白质积聚于脑中而发病的变性性痴呆症。后者有β-淀粉样蛋白(aβ)和tau积聚的阿尔茨海默病(ad)、tau或tdp-43积聚的额颞叶痴呆症(ftd)、α-突触核蛋白积聚的路易小体痴呆症(dlb)等。认为这些蛋白质在脑内形成寡聚体,并损害神经细胞的功能而发病。基于该想法,正在开发具有抑制这些蛋白质的产生、抑制寡聚体的形成、将凝集的蛋白质从脑中除去等作用的药来作为变性性痴呆症的治疗药。
例如,对于ad,已经开发了与aβ的产生有关的酶(β-分泌酶或γ-分泌酶)的抑制药、将aβ从脑中除去的aβ疫苗、aβ抗体等,供于临床试验。但是,迄今为止,由于出现出乎意料的副作用或没有产生所期待的药效等理由,其大部分在临床上失败。
另一方面,作为抗生素而众所周知的利福平因其抗菌作用,一直以来被用作经口药。进而,还已知利福平具有自由基除去作用,作为其作用之一,报道有与抑制aβ凝集反应有关(参照非专利文献1)。
现有技术文献
非专利文献
非专利文献1:tomiyama,t.外6名著、“inhibitionofamyloidbetaproteinaggregationandneurotoxicitybyrifampicin.itspossiblefunctionasahydroxylradicalscavenger.”、jbiolchem、1996年、vol.271、p.6839-6844
技术实现要素:
发明所要解决的问题
认为以ad为对象的临床试验中的aβ标靶药(aβ产生酶的抑制药、aβ疫苗、aβ抗体等)的失败原因在于,如果不考虑副作用的问题,则在于给药时期过晚。即,认为除去aβ如果不是在神经细胞开始死亡的痴呆症发病前则没有意义。因此,aβ标靶药的作用应该不是在于治疗而在于预防。需要说明的是,报道有即使通过tau标靶药中最初进入临床试验的tau凝集抑制药,在以ad为对象的临床试验中,也没有看到认知功能改善作用。认为该结果也显示出,即使是tau标靶药,在痴呆症发病后给药也过晚。
但是,现在许多开发中的治疗药并不是在预防性给药的前提下开发的,在费用、副作用、给药法等方面存在问题。另一方面,为了预防痴呆症而投药时,由于发病时间不清楚,因此,需要设想相当长的期间作为投药期间。
另一方面,本发明人发现:利福平在活体外会抑制aβ、tau、α-突触核蛋白的寡聚体形成;并且在对积聚aβ的ad的模型小鼠或积聚tau的ftd的模型小鼠经口给药时,可抑制这些蛋白质寡聚体的脑内积聚,使小鼠的认知功能恢复。因此,本发明人研究了利用利福平的这种作用,以将一直以来用作抗生素的经口药利福平重新定位为痴呆症预防药,使其实用化。
但是,进行这样的研究的结果面临如下问题:利福平引起的肝损害或药物相互作用等副作用是严重的,因此,不能在以利福平为预防药的前提下长期服用。在此的药物相互作用是指以下现象:利福平在肝细胞中诱导与药物代谢有关的细胞色素p450(cyp)和p-糖蛋白,从而使同时服用的其它药的效果减弱。
在此,经口摄取利福平时,从小肠被吸收,经过门静脉而被运输至肝脏。通常,所吸收的药的大部分在肝脏中被分解、失活,仍旧保持活性而进入大循环的是所给药的药的极少一部分(将其称为首过效应)。进而,在血液和脑之间存在限制物质交换的血脑屏障(bbb),血液中的利福平中的进入脑的利福平为其另外极少一部分(数%~数十%左右)。认为在迄今为止的利福平的投药中,通过摄取预先补充了因首过效应及bbb而失去的量的相应量,其结果,确保了显现利福平的药效所需要的脑内浓度。而且,认为在药通过肝脏时产生了利福平的副作用。
因此,本发明的目的在于,提供一种通过提高利福平向脑的直接转移性、抑制向肝脏的首次通过,从而能够长期给药的利福平的投药技术。
用于解决问题的技术方案
本发明人深入研究的结果发现,通过经鼻给药利福平,可解决上述课题。本发明是基于上述的见解进一步重复进行研究而完成的。
即,本发明提供以下所示的方式的发明。
方案1.一种经鼻给药用医药组合物,其含有选自利福平、其衍生物、及它们的盐中的利福平类作为有效成分,用于痴呆症的预防或治疗。
方案2.根据方案1所述的经鼻给药用医药组合物,其中,所述利福平类的有效给药量为0.15mg/kg/天以上且3.75mg/kg/天以下。
方案3.根据方案1或2所述的经鼻给药用医药组合物,其用于痴呆症的预防。
方案4.根据方案1~3中任一项所述的经鼻给药用医药组合物,其中,所述痴呆症为阿尔茨海默病。
方案5.一种利福平类的用途,其用于制造用于痴呆症的预防或治疗的经鼻给药用医药组合物,且选自利福平、其衍生物、及它们的盐中利福平。
方案6.一种痴呆症的治疗方法,其包含以下工序:对痴呆症患者经鼻给药有效量的选自利福平、其衍生物、及它们的盐中的利福平类。
方案7.一种痴呆症的预防方法,其包含以下工序:对痴呆症发病风险高的未发病者经鼻给药有效量的选自利福平、其衍生物、及它们的盐中的利福平类。
发明效果
根据本发明的医药组合物,提供一种投药技术,其通过制备利福平作为经鼻给药药剂,从而实现非侵入、高药效及低副作用,由此能够长期给药,因此,不仅可以应用于痴呆症的治疗,而且也可以应用于痴呆症的预防。
附图说明
图1是实验例1中进行的行动试验的结果,显示利福平给药对小鼠的认知功能的利福平效果。
图2是实验例1中进行的免疫染色的结果,显示利福平对aβ寡聚体、突触素的利福平效果。
图3显示基于图2的免疫染色的aβ寡聚体的定量结果。
图4显示基于图2的免疫染色的突触素的定量结果。
图5是实验例1中进行的免疫染色的结果,显示利福平对磷酸化tau的利福平效果。
图6显示基于图5的免疫染色的磷酸化tau的定量结果。
具体实施方式
本发明的医药组合物的特征在于,含有选自利福平、其衍生物、及它们的盐中的利福平类作为有效成分,用于痴呆症的预防或治疗、且进行经鼻给药。
[利福平类]
本发明的医药组合物含有选自利福平、其衍生物、及它们的盐中的利福平类作为有效成分。利福平类具有抑制作为变性性痴呆症的阿尔茨海默病、额颞叶痴呆症、路易小体痴呆症等的原因蛋白质即β-淀粉样蛋白、tau、α-突触核蛋白的寡聚体形成的作用。认为利福平类具有萘氢醌或萘醌结构,该结构有助于作为利福平的自由基清除剂的作用。
利福平通常为下述式(i)表示的化合物。
作为利福平的衍生物,只要是具有萘氢醌或萘醌结构且在药学上允许的物质,就没有特别限定,例如可举出3-甲酰基利福霉素sv、利福霉素s、利福霉素b、利福霉素sv、主要活性代谢物25-去乙酰基利福平等。在利福平衍生物中,从抑制因长期给药导致的耐药菌的诱导的观点考虑,优选不具有担负抗生素活性的1,4-二羟基萘结构的3位取代基的衍生物、例如利福霉素sv。这些利福平的衍生物可以单独使用1种,另外,也可以将2种以上组合而使用。
作为利福平的盐,只要是与利福平或利福平的衍生物形成盐且在药学上允许的化合物,就没有特别限定。可举出例如:碱金属(钾、钠等)的盐、碱土金属(钙、镁等)的盐、铵盐、药学上允许的有机胺(四甲基铵、三乙基胺、甲基胺、二甲基胺、环戊胺、苄胺、苯乙基胺、哌啶、单乙醇胺、二乙醇胺、三(羟基甲基)氨基甲烷、赖氨酸、精氨酸、n-甲基-d-葡糖胺等)的盐、无机酸盐(盐酸盐、氢溴酸盐、氢碘酸盐、硫酸盐、磷酸盐、硝酸盐等)、有机酸盐(醋酸盐、乳酸盐、酒石酸盐、苯甲酸盐、柠檬酸盐、甲磺酸盐、乙磺酸盐、苯磺酸盐、甲苯磺酸盐、羟乙磺酸盐、葡萄糖醛酸、葡萄糖酸盐等)。这些盐可以单独使用1种,另外,也可以将2种以上组合而使用。
作为利福平类,可以从利福平、利福平的盐、利福平的衍生物、利福平的衍生物的盐中选择一种而使用,也可以将两种以上组合而使用。
上述的利福平类中,优选举出利福平及利福霉素sv。
[剂型]
本发明的医药组合物作为经鼻给药剂被制备。经鼻给药剂将上述的利福平类作为有效成分,通过其自身公知的方法而被制剂化,可以适当混合药理学上所允许的基剂和/或添加物。
作为药理学上所允许的基剂和/或添加物,例如可举出赋形剂、粘稠剂、润滑剂、粘结剂、崩解剂、溶剂、增溶剂、悬浮剂、乳化剂、张度剂、缓冲剂、安慰剂(無痛化剤)、稳定剂等。另外,根据需要可以含有保存剂(防腐剂)、ph调节剂、清凉剂、抗氧化剂、湿润剂、粘合剂、矫臭剂等添加物。
作为赋形剂,例如可举出乳糖、白糖、d-甘露醇、淀粉、玉米淀粉、结晶纤维素、轻质无水硅酸等。作为粘稠剂,可举出例如:甘油、聚乙二醇等多元醇、甲基纤维素、羧甲基纤维素、羟基丙基甲基纤维素等纤维素类、聚乙烯醇、聚乙烯基吡咯烷酮、羧基乙烯基聚合物、羧甲基纤维素钠、甲基纤维素、羟基甲基纤维素、羟基乙基纤维素、羟基丙基纤维素等亲水性高分子、海藻酸钠、硫酸软骨素、环糊精、d-α-生育酚聚乙二醇1000琥珀酸酯(d-α-トコフェリルポリエチレングリコール1000コハク酸)、聚乙二醇等。作为润滑剂,可举出例如:硬脂酸镁、硬脂酸钙、滑石、胶态二氧化硅等。作为粘结剂,可举出例如:结晶纤维素、白糖、d-甘露醇、糊精、羟基丙基纤维素、羟基丙基甲基纤维素、聚乙烯基吡咯烷酮、淀粉、蔗糖、明胶、甲基纤维素、羧甲基纤维素钠等。作为崩解剂,可举出例如:淀粉、羧甲基纤维素、羧甲基纤维素钙、交联羧甲基纤维素钠、羧甲基淀粉钠、l-羟基丙基纤维素等。作为溶剂,可举出例如:水、乙醇、异丙醇、丙酮、丙二醇、聚乙二醇、胡麻油、玉米油等。作为增溶剂,可举出例如:甲基纤维素、羧甲基纤维素、羟基丙基甲基纤维素等纤维素类;聚乙二醇、丙二醇、d-甘露醇、苯甲酸苄酯、乙醇、三氨基甲烷、胆固醇、三乙醇胺、碳酸钠、柠檬酸钠、聚乙烯基吡咯烷酮、聚乙二醇等。作为悬浮剂,可举出例如:硬脂基三乙醇胺、月桂基硫酸钠、月桂基氨基丙酸、卵磷脂、苯扎氯铵、苄索氯铵、单硬脂酸甘油、聚氧乙烯氢化蓖麻油、聚山梨酯等表面活性剂、甘油、聚乙二醇等多元醇、山梨糖醇、甘露醇、蔗糖等糖类、甲基纤维素、羧甲基纤维素、羟基丙基甲基纤维素等纤维素类、聚乙烯醇、聚乙烯基吡咯烷酮、羧基乙烯基聚合物、羧甲基纤维素钠、甲基纤维素、羟基甲基纤维素、羟基乙基纤维素、羟基丙基纤维素等亲水性高分子、硫酸软骨素等。作为张度剂,可举出例如:葡萄糖、d-山梨糖醇、氯化钠、甘油、d-甘露醇、氯化钾、浓甘油、丙二醇、蔗糖等。作为缓冲剂,可举出例如:磷酸盐(磷酸氢钠、磷酸二氢钠等)、硼酸、硼砂、醋酸盐(醋酸钠等)、碳酸盐(碳酸钠、碳酸钙、碳酸钾等)、柠檬酸、l-谷氨酸钠等。作为安慰剂,可举出例如:苄醇、氯丁醇、丙二醇、氨基苯甲酸乙酯、利多卡因等。作为稳定剂,可举出例如:亚硫酸钠、亚硫酸氢钠、偏亚硫酸氢钠(メタ亜硫酸水素ナトリウム)、硫代硫酸钠、雕白粉、硫代甘油、巯基乙酸、硫代乳酸、半胱氨酸、谷胱甘肽、硫代醋酸、蛋氨酸、硫代山梨糖醇、硫代葡萄糖、硫脲等硫化合物、硼酸、硼砂、磷酸、偏磷酸、碳酸钠、碳酸氢钠等无机酸及其盐类、甲酸、草酸、酒石酸、柠檬酸、乙二胺四乙酸等有机酸及其盐类(乙二胺四乙酸钠等)、乙酰胺、二乙基乙酰胺、烟酰胺、尿素、巴比妥等酰胺;尿素衍生物、乙二醇、丙二醇、甘油、聚乙二醇、葡萄糖、抗坏血酸等多元醇、糖类、苯酚、百里酚、醌、香豆酮、异香豆酮等酚类;二丁基羟基甲苯、甘氨酸、谷氨酸、赖氨酸、苯基丙氨酸、酪蛋白、麻仁球蛋白等氨基酸、蛋白质等。作为乳化剂,可举出例如:甘油酯(单油酸甘油酯)、皂角苷(槐树皂角苷、皂树皮提取物、大豆皂角苷等)、蔗糖脂肪酸酯、卵磷脂(植物卵磷脂、蛋黄卵磷脂、大豆卵磷脂等)、多元醇(油醇、硬脂基醇、十六烷基醇等)、脂肪酯(肉豆蔻酸辛基十二烷基酯等)、中链脂肪酸甘油三酯(mct)、各种表面活性剂(烷基苯磺酸盐型乳化剂、苯扎氯铵、山梨糖醇酐倍半油酸酯、十二烷基苯磺酸等)、三乙醇胺等。作为保存剂(防腐剂),可举出例如:对羟基苯甲酸丙酯、对羟基苯甲酸丁酯等对羟基苯甲酸酯类(パラオキシ安息香酸エステル類)、羟苯甲酯、羟苯乙酯、羟苯丙酯、羟苯丁酯等羟苯酯类(パラベン類)、苯扎氯铵、苄索氯铵、葡萄糖酸氯己啶、氯化十六烷基吡啶
本发明的医药组合物可以为液体剂、固体剂中的任一种,优选举出液体剂。在设为液体剂的情况下,可以通过将利福平类和根据需要的溶剂、增溶剂、悬浮剂、张度剂、缓冲剂、安慰剂等混合并进行溶解、悬浮或乳化而制造。也优选进一步加入粘稠剂而提高粘性并赋予滞留性。在设为固体剂的情况下,可以通过将利福平类和赋形剂、粘结剂、崩解剂或其它适当的添加剂均匀地混合,利用适当的造粒法得到造粒物,进一步根据需要通过干燥制成粉末或颗粒状而制造。
本发明的医药组合物中,作为利福平的含量,只要制备为经鼻给药药剂,则没有特别限定,以可以根据后述的用量给药的方式适当调整。例如,作为本发明的医药组合物中的利福平的含量,可举出0.4w/v%以上、优选0.5w/v%以上。另外,从以少的给药次数有效率地给药有效量的观点考虑,本发明的医药组合物中的利福平的含量可以为2w/v%以上、2.5w/v%以上、5w/v%以上、或30w/v%以上。进而,作为本发明的医药组合物中的利福平的含量,可举出95w/v%以下,从良好地得到经鼻给药药剂的喷雾性等的观点考虑,可举出85w/v%以下、或50w/v%以下,作为本发明的医药组合物中的利福平含量的具体范围,可举出例如:0.4~95w/v%、0.4~85w/v%、0.4~50w/v%、0.5~95w/v%、0.5~85w/v%、0.5~50w/v%、2~95w/v%、2~85w/v%、2~50w/v%、2.5~95w/v%、2.5~85w/v%、2.5~50w/v%、5~95w/v%、5~85w/v%、5~50w/v%、30~95w/v%、30~85w/v%、30~50w/v%。
本发明的医药组合物可以填充于经鼻给药用的容器而使用。经鼻给药用的容器可以适当使用市售的容器。
[用量及用法]
本发明的医药组合物被制备为经鼻给药用,因此,与经口给药相比,能够以少的用量产生经口给药以上的药效,同时也可以减轻肝损害的副作用。因此,与作为抗生素进行经口给药的情况相比,该情况能够长期间连续地给药少的用量。
作为本发明的医药组合物的利福平对人的有效给药量,从显现药效的观点考虑,可举出作为抗生素进行经口给药时的用量(例如7.5~10mg/kg/天)的例如1/50以上、优选1/25以上、更优选1/10以上、进一步优选1/7.5以上,从抑制副作用的观点考虑,可举出作为抗生素进行经口给药时的用量(例如7.5~10mg/kg/天)的例如1/2以下、优选1/3以下、更优选1/3.75以下。作为利福平对人的有效给药量的具体范围,可举出作为抗生素进行经口给药时的用量(例如7.5~10mg/kg/天)的例如1/50~1/2、1/50~1/3、1/50~1/3.75、1/25~1/2、1/25~1/3、1/25~1/3.75、1/10~1/2、1/10~1/3、1/10~1/3.75、1/7.5~1/2、1/7.5~1/3、1/7.5~1/3.75,特别举出例如:1/50以上且1/2以下、优选1/25以上且1/3以下、更优选1/10以上且1/3以下、进一步优选1/7.5以上且1/3.75以下。
作为更具体的本发明的医药组合物的利福平的有效给药量,从显现药效的观点考虑,可举出例如0.15mg/kg/天以上、优选0.3mg/kg/天以上、更优选0.75mg/kg/天以上、进一步优选1mg/kg/天以上,从抑制副作用的观点考虑,可举出例如3.75mg/kg/天以下、优选2.5mg/kg/天以下、更优选2mg/kg/天以下。作为本发明的医药组合物的利福平的有效给药量的具体范围,可举出:0.15~3.75mg/kg/天、0.15~2.5mg/kg/天、0.15~2mg/kg/天、0.3~3.75mg/kg/天、0.3~2.5mg/kg/天、0.3~2mg/kg/天、0.75~3.75mg/kg/天、0.75~2.5mg/kg/天、0.75~2mg/kg/天、1~3.75mg/kg/天、1~2.5mg/kg/天、1~2mg/kg/天,特别举出例如:0.15~3.75mg/kg/天、优选0.3~2.5mg/kg/天、更优选0.75~2.5mg/kg/天、进一步优选1~2mg/kg/天(特别优选1.67mg/kg/天)。
本发明的医药组合物可以以少的用量进行给药,因此,适于连续给药。因此,可以长期地给药。例如,可以进行6个月以上、例如6个月~3年的给药。另外,作为给药间隔,可举出隔天、或每1周1~2次。
[给药对象]
本发明的医药组合物可以用于痴呆症的预防及痴呆症的治疗。优选可以用于痴呆症的预防。痴呆症可举出变性性痴呆症及脑血管性痴呆症,优选举出变性性痴呆症。作为变性性痴呆症,可举出通过β-淀粉样蛋白(aβ)、tau、tdp-43、α-突触核蛋白等痴呆症原因蛋白质积聚而发病的痴呆症,具体而言,可举出:β-淀粉样蛋白(aβ)和tau积聚的阿尔茨海默病(ad)、tau或tdp-43积聚的额颞叶痴呆症(ftd)、α-突触核蛋白积聚的路易小体痴呆症(dlb)等,优选举出阿尔茨海默病(ad)。
将本发明的医药组合物用于痴呆症的预防的情况下,只要是发病风险高的未发病者,就没有特别限定。作为发病风险高的未发病者,可举出老年斑阳性的健康人、家族性阿尔茨海默病系的家族等。用于痴呆症的治疗的情况下,作为给药对象,只要是完成了痴呆症的诊断、需要阻止痴呆症的症状的进行的患者,就没有特别限定。
[药理作用]
在鼻腔上部的鼻粘膜产生嗅上皮神经元的树状突起,位于其细胞表面的嗅觉受体中得到的臭味信息沿着嗅上皮神经元的轴突而被送到脑的嗅球。在鼻粘膜和嗅上皮神经元之间不存在血脑屏障(bbb)。虽然在束住嗅上皮神经元的轴突的神经束的周围存在脑脊髄液,但在此也不存在阻碍血液和脑脊髄液之间的物质交换的血脑脊液屏障(bcsfb)。因此,通过本发明的医药组合物的经鼻给药,到达鼻粘膜的有效成分利福平类可以不受到因bbb及bcsfb所致的损害而进入嗅上皮神经元或脑脊髄液,并转移至脑内。
这样,根据本发明的医药组合物,利福平类向脑的直接转移性得以提高,因此,可以抑制向肝脏的首次通过。因此,本发明的医药组合物不仅在给药形式方面为非侵入的,而且取得因提高了向脑的直接转移性所致的高药效、及因抑制向肝脏的首次通过所致的低副作用。
到达脑的利福平类在变性性痴呆症的情况下,招致抑制β-淀粉样蛋白(aβ)、tau、tdp-43、α-突触核蛋白等痴呆症原因蛋白质的寡聚体的形成或凝集、或使所形成或凝集的痴呆症原因蛋白质的寡聚体消失。由此,带来痴呆症的发病延迟化,或发病的痴呆症的症状的改善(例如通过使突触复活而恢复记忆障碍)。或者,到达脑的利福平类在脑血管性痴呆症的情况下,通过经由自由基清除剂作用的神经保护作用而改善脑血管障碍。由此,带来痴呆症的症状的改善。
实施例
以下,示出实施例,更具体地说明本发明,但本发明并不限定于这些实施例。
[实验例]
在本实验例中,以表1所示的用量及用法向表1所示的小鼠每天给药含有利福平或不含有利福平的给药组合物1个月。
(给药对象)
准备11月龄的雄性apposk小鼠(tomiyamaetal.jneurosci.2010;30:4845-56)。apposk小鼠的体重约为30g。将apposk小鼠60只每12只分成a-e的5组。另外,准备同月龄的野生型小鼠(非转基因同窝鼠(non-tglittermate))12只。需要说明的是,apposk小鼠为淀粉样前体蛋白(app)转基因小鼠(阿尔茨海默病模型),显示β-淀粉样蛋白(aβ)的积聚。
(给药组合物)
将利福平药(rfp;sigma-aldrich,rifampicin≥97%(hplc),粉末,别名:3-(4-甲基哌嗪基亚氨基甲基)利福霉素sv,利福霉素amp,利福平,r3501)以成为表1的比较例1、实施例1~2、参考例3所示的容量的配合量悬浮在0.5w/v%的羧甲基纤维素(cmc,sigma-aldrich,羧甲基纤维素钠盐低粘度(carboxymethylcellulosesodiumsaltlowviscosity),c5678)溶液中,制备给药组合物。
(给药方法)
经口给药使用啮齿类用经口探针,经鼻给药使用pipetman(白吸头),皮下给药使用注射器,全部在无麻醉下进行。
[表1]
(结果1-行动试验)
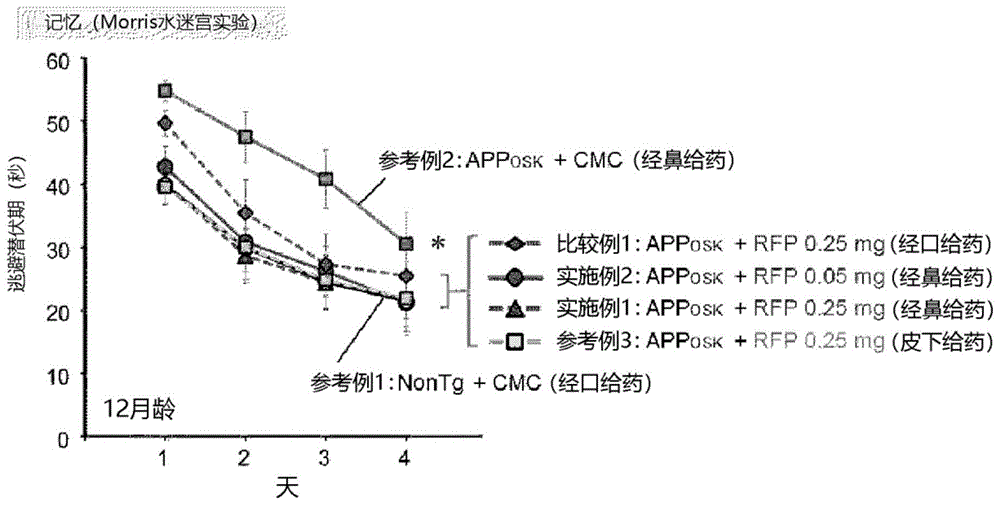
将给药结束后(12月龄)的小鼠供于行动试验,比较利福平对小鼠的认知功能的效果。行动试验是按照umedaetal.brain2016;139:1568-86的方法,利用morris水迷宫测定小鼠的空间参考记忆而进行的。需要说明的是,供于行动试验的小鼠除了在给药期间死亡的小鼠之外,为12只(比较例1、实施例1、实施例2)、11只(参考例1、参考例2、参考例3)。
将行动试验的结果示于图1。在经口给药(oral)、经鼻给药(intranasal)、皮下给药(subcutaneous)的任一种情况下,均确认了apposk小鼠的记忆障碍的改善。但是,通过经口给药(比较例1)时,其改善效果不完全。与此相对,通过经鼻给药(实施例1及实施例2)和皮下给药(参考例3)时,其改善效果达到与野生型小鼠同水平。进而,确认了通过经鼻给药时,即使在将用量下降至1/5(0.05mg/天)的情况(实施例2)下,改善效果也比通过经口给药的情况(比较例1)高。
(结果2-肝功能障碍)
从行动试验结束后的小鼠中采血,将血清进行分离而制备血清试样。进行血清试样的肝酶ast(got)及alt(gpt)的测定,比较利用利福平的肝功能障碍的程度。
将肝酶的测定结果示于表2。与cmc给药apposk小鼠(参考例2)相比,在rfp经口给药小鼠(比较例1)中ast显著地上升,启示了肝毒性。与此相对,在经鼻给药小鼠(实施例1、实施例2)中,没有看到比较例1那样的上升。需要说明的是,认为经鼻给药小鼠(实施例1、实施例2)中的ast的稍微上升是鼻腔内给药的药的一部分流向咽喉并由小肠吸收而引起的。与小鼠不同,在对人的经鼻给药中,认为几乎不会有这种误饮。在皮下给药(参考例3)中没有看到ast的上升。关于alt,通过任一种给药法,均没有看到显著的变化。
[表2]
ast、alt的数值用mean±sem(iu/l)表示。
*与non-tg小鼠、cmc给药tg小鼠及皮下给药tg小鼠的ast值各自的组间差的p值(根据tukey-kramertest)为p<0.05。
就c57bl6小鼠中的正常值而言,ast为68±24、alt为30±8。
(结果3-免疫组织染色)
从行动试验结束后的小鼠中取出脑,通过免疫组织化学染色,比较利福平对aβ寡聚体、突触素、磷酸化tau的效果。
按照umedaetal.brain2016;139:1568-86中记载的步骤,通过免疫组织化学染色法,进行aβ寡聚体(认为aβ寡聚体引起tau的磷酸化及突触素的减少)、突触素(突触的标记蛋白)、及磷酸化tau的染色。在aβ寡聚体的染色中使用11a1抗体(株式会社免疫生物研究所制),在突触素的染色中使用svp-38抗体(sigma社制),在磷酸化tau的染色中使用小鼠单克隆phf-1抗体(抗p-ser396/404-tau抗体、由阿尔伯特爱因斯坦医学院的peterdavies博士提供)。染色后,使用nihimage-j,进行aβ寡聚体、突触素、及磷酸化tau的定量。
将aβ寡聚体(aβoligomers)及突触素(synaptophysin)的免疫染色后的组织的照片示于图2。图2中,上段表示海马ca3组织,下段表示海马ca2/3组织。rfp的经口给药(比较例1)、经鼻给药(实施例1、实施例2)、皮下给药(参考例3)中的任一种情况下,均是积聚于脑的aβ寡聚体减少,减少的突触素恢复。
将由图2的免疫染色结果得到的aβ寡聚体的定量结果示于图3。在rfp的经口给药(比较例1)、经鼻给药(实施例1、实施例2)、皮下给药(参考例3)的任一种中,aβ寡聚体均减少至与野生型小鼠(参考例1)至少同水平。其中,以相同的用量进行比较时,aβ寡聚体的减少效果在经鼻给药(实施例1)时最高。
将由图2的免疫染色结果得到的突触素的定量结果示于图4。在rfp的经口给药(比较例1)、经鼻给药(实施例1、实施例2)、皮下给药(参考例3)的任一种中,海马的突触素均显示出恢复的倾向。其中,经口给药(比较例1)产生的效果弱,另一方面,通过经鼻给药(实施例1、实施例2)、皮下给药(参考例3)时,恢复至与野生型小鼠(参考例1)同水平。进而,确认了与行动试验的结果同样地,在经鼻给药的情况、将用量下降至1/5(0.05mg/天)的情况(实施例2)下,其效果均比经口给药(比较例1)高。
将磷酸化tau(phosphorylatedtau)的免疫染色后的组织的照片示于图5。图5表示海马ca2/3组织。rfp的经口给药(比较例1)、经鼻给药(实施例1、实施例2)、皮下给药(参考例3)的任一种情况下,积聚于脑的磷酸化tau均减少。
将由图5的免疫染色结果得到的磷酸化tau的定量结果示于图6。在rfp的经口给药(比较例1)、经鼻给药(实施例1、实施例2)、皮下给药(参考例3)的任一种中,海马的磷酸化tau均显示出减少倾向。其中,经口给药(比较例1)产生的效果弱,另一方面,经鼻给药(实施例1、实施例2)、皮下给药(参考例3)产生的效果高。进而,以相同的用量进行比较时,磷酸化tau的减少效果在经鼻给药(实施例1)时最高。
由以上的结果证实:在利福平的给药中,经鼻给药在药效高且副作用低方面与经口给药相比优异,且在非侵入性的方面与皮下给药相比优异。另外,产生这种效果的对小鼠的1个月的给药期间就人而言相当于3.3年左右。因此,利福平的经鼻给药适于长期的给药,因此显示了不仅适于痴呆症的治疗,而且也适于痴呆症的预防。在本实施例中,示出了相对于小鼠(体重约30g)为0.05mg/只/天(1.67mg/kg/天)及0.25mg/只/天(8.33mg/kg/天)的有效给药量作为经鼻给药量,由得到的结果,以这些给药量的1成左右的给药量,也可以期待效果。另外,认为也可以更长期的给药,因此,考虑这种更长期的给药时,以更少的给药量(例如0.15mg/kg/天)也可以期待效果。另一方面,鉴于利福平对人的经口给药量在处方中为450~600mg/60kg/天(7.5~10mg/kg/天),并且在上述的实施例中即使经鼻给药为经口给药的1/5的量这样的少量也显现效果,在对人的给药中,认为以目前的1/2的给药量(例如3.75mg/kg/天)也当然是有效的。综上所述,在对人的给药中,可以将0.15~3.75mg/kg/天设为有效给药量。
- 还没有人留言评论。精彩留言会获得点赞!