免疫受体条形码错误校正的制作方法
相关申请的交叉引用
本申请要求于2017年9月25日提交的美国临时申请号62/562,978的优先权。相关申请的内容通过引用以其全文明确地并入本文。
序列表的引用
本申请是连同电子格式的序列表一起提交的。序列表被提供为题为sequence_listing_bdcri_035wo.txt的文件,创建于2018年9月21日,大小是32千字节。将电子格式的序列表的信息通过引用以其全文并入本文。
发明背景
本披露总体上涉及分子条形码化的领域,并更具体地涉及使用分子标记来校正取代错误和非取代错误。
背景技术:
诸如条形码化(包括随机条形码化)等方法和技术对于细胞分析是有用的,特别是使用例如逆转录、聚合酶链式反应(pcr)扩增、和下一代测序(ngs)来解密基因表达谱以确定细胞的状态。然而,这些方法和技术可能引入错误(诸如取代错误(例如,涉及一个或多个碱基取代的取代错误))和非取代错误(例如,引物交叉错误和pcr嵌合体错误),如果未校正,其可导致过高估计的分子计数。因此,需要能够校正各种错误的方法和技术以获得准确的分子计数。
技术实现要素:
本文披露的内容包括用于确定靶的出现的方法。在一些实施例中,所述方法包括:(a)使用多个条形码(例如,随机条形码)使多个靶条形码化(例如,随机条形码化)以创建多个经条形码化的靶(例如,经随机条形码化的靶),其中,所述多个条形码中的每一个包括细胞标记和分子标记,其中,所述多个条形码中的至少两个条形码的分子标记包括不同的分子标记序列,并且其中所述多个条形码中的至少两个条形码包括具有相同的细胞标记序列的细胞标记;(b)获得所述经条形码化的靶的测序数据;以及(c)针对所述多个靶中的至少一个靶:(i)鉴定所述测序数据中的所述靶的推定序列;(ii)对与(i)中鉴定的所述测序数据中的所述靶的推定序列相关联的分子标记序列的出现进行计数;(iii)鉴定所述靶的推定序列的簇;(iv)使用(iii)中鉴定的所述靶的推定序列的簇对获得的测序数据进行折叠;(v)鉴定与所述靶的推定序列相关联的所述分子标记序列的簇;(vi)使用(v)中鉴定的所述分子标记序列的簇对所述测序数据进行折叠;(vii)鉴定组合序列的簇,其中,每个组合序列包括所述靶的序列中的序列以及所述分子标记序列中的相关联的分子标记序列;(viii)使用(vii)中鉴定的所述组合序列的簇对所述测序数据进行折叠;(ix)鉴定与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列,其中,与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列的出现小于不与所述靶的一个或多个嵌合序列相对应的所述靶的剩余一个或多个推定序列的出现;(x)从所述测序数据中去除(ix)中鉴定的与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列;以及(xi)估计所述靶的出现,其中,在(iv)、(vi)和(viii)对所述测序数据进行折叠并且在(x)中去除与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列后,估计的所述靶的出现与(ii)中计数的分子标记序列的数量相关。
在一些实施例中,所述多个靶包括细胞的整个转录组的靶。所述多个靶可以包括基因。所述基因可以包括对免疫受体进行编码的可变序列,诸如可变(v)区、多样性(d)区、连接(j)区或其任何组合。所述基因可以是对t细胞受体进行编码的基因。所述靶的推定序列可以彼此相差至少一个核苷酸。
在一些实施例中,鉴定所述靶的推定序列的簇包括使用定向邻接鉴定所述靶的推定序列的簇。簇内所述靶的推定序列可以在彼此的第一预定定向邻接阈值内。所述第一定向邻接阈值可以是汉明距离(hammingdistance)。所述簇内所述靶的推定序列可以包括一个或多个亲本序列以及所述一个或多个亲本序列的一个或多个子序列,并且其中,所述亲本序列的出现大于或等于第一预定定向邻接出现阈值。所述第一预定定向邻接出现阈值可以是小于一的子序列的出现的两倍。
在一些实施例中,使用(iii)中鉴定的所述靶的推定序列的簇对在(b)中获得的所述测序数据进行折叠包括:将所述一个或多个子序列中的子序列的出现归因于所述子序列的亲本序列。
在一些实施例中,鉴定与所述靶的推定序列相关联的所述分子标记序列的簇包括使用定向邻接鉴定与所述靶的推定序列相关联的所述分子标记序列的簇。簇内所述靶的分子标记序列可以在彼此的第二预定定向邻接阈值内。所述第二定向邻接阈值可以是汉明距离。所述簇内所述靶的推定分子标记序列可以包括一个或多个亲本分子标记序列以及所述一个或多个亲本分子标记序列的一个或多个子分子标记序列,并且其中,所述亲本分子标记序列的出现大于或等于第二预定定向邻接出现阈值。所述第二预定定向邻接出现阈值可以是小于一的子分子标记序列的出现的两倍。
在一些实施例中,使用(v)中鉴定的与所述靶的序列相关联的所述分子标记序列的簇对所述测序数据进行折叠包括:将所述一个或多个子分子标记序列中的子分子标记序列的出现归因于所述子分子标记序列的亲本分子标记。
在一些实施例中,鉴定所述组合序列的簇包括使用定向邻接来鉴定所述组合序列的簇。簇内的组合序列可以在彼此的第三预定定向邻接阈值内。所述第三定向邻接阈值可以是汉明距离。所述簇内的所述组合序列可以包括一个或多个亲本组合序列以及所述一个或多个亲本组合序列的一个或多个子组合序列,并且其中,所述亲本组合序列的出现大于或等于第三预定定向邻接出现阈值。所述第三预定定向邻接出现阈值可以是小于一的子组合序列的出现的两倍。
在一些实施例中,使用(vii)中鉴定的所述组合序列的簇对所述测序数据进行折叠包括:将所述一个或多个子组合序列中的子组合序列的出现归因于所述子组合序列的亲本组合序列。
在一些实施例中,鉴定与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列:鉴定与所述多个分子序列中的一个分子标记序列相关联的所述靶的推定序列;鉴定与所述一个分子标记序列相关联的所述靶的推定序列中的推定序列,所述一个分子标记序列的出现小于与所述靶的一个或多个嵌合序列中的嵌合序列相对应的嵌合出现阈值。嵌合出现阈值的值可以是与一个分子标记序列相关联的所述靶的推定序列中的推定序列的出现,所述出现大于所述靶的推定序列中的任何其他序列的出现。
在一些实施例中,所述方法进一步包括:在(iv)、(vi)和(viii)中对所述测序数据进行折叠并且在(x)中去除与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列后调整所述测序数据。在(iv)、(vi)和(viii)对所述测序数据进行折叠并且在(x)中去除与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列后调整所述测序数据可以包括:在(iv)、(vi)和(viii)中对所述测序数据进行折叠并且在(x)中去除与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列后,将与所述靶的推定序列相关联的分子标记序列阈值化,以确定与(b)中计数的所述测序数据中的所述靶的序列相关联的信号分子标记序列和噪音分子标记序列。将与所述靶的推定序列相关联的所述分子标记序列阈值化可以包括对所述靶的所述分子标记序列进行统计分析。进行所述统计分析可以包括:将与所述靶的推定序列相关联的所述分子标记序列及其出现拟合至两个负二项分布;使用所述两个负二项分布来确定信号分子标记序列的出现n;以及在(iv)、(vi)和(viii)对所述测序数据进行折叠并且在(x)中去除与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列后,从在(b)中获得的测序数据去除所述噪音分子标记序列,其中,所述噪音分子标记序列包括其出现小于第n个最丰富分子标记的出现的分子标记序列,并且其中所述信号分子标记序列包括其出现大于或等于第n个最丰富分子标记的出现的分子标记序列。这两个负二项分布可以包括与所述信号分子标记序列相对应的第一负二项分布以及与所述噪音分子标记序列相对应的第二负二项分布。
本文披露的是用于确定靶的出现的方法。在一些实施例中,所述方法包括:(a)接收多个靶的测序数据,其中,所述测序数据包括所述多个靶中的靶的推定序列以及与所述测序数据中的所述靶的序列相关联的分子标记序列的出现;(b)对所述靶的推定序列进行折叠;(c)对与所述靶的推定序列相关联的分子标记序列进行折叠;以及(d)估计所述靶的出现,其中在(b)中对所述靶的推定序列的出现进行折叠并且对在(c)中确定的噪音分子标记序列的出现进行折叠之后,估计的所述靶的出现与跟所述测序数据中的所述靶的推定序列相关联的分子标记序列的出现相关。
在一些实施例中,所述方法包括:鉴定所述测序数据中的所述靶的序列;以及对与所述测序数据中的所述靶的序列相关联的分子标记序列的出现进行计数。
在一些实施例中,所述方法包括:对组合序列的簇进行折叠,其中,每个组合序列包括所述靶的序列中的序列以及所述分子标记序列中的相关联的分子标记序列,其中,在对所述组合序列的出现进行折叠后,估计的所述靶的出现与跟所述测序数据中的所述靶的序列相关联的分子标记序列的出现相关。对所述组合序列的簇进行折叠可以包括:使用定向邻接对所述组合序列的簇进行折叠。使用定向邻接对所述组合序列的簇进行折叠可以包括:使用定向邻接鉴定所述组合序列的簇;以及使用鉴定的所述组合序列的簇对所述测序数据进行折叠。对所述靶的推定序列进行折叠包括:使用定向邻接对所述靶的推定序列进行折叠。使用定向邻接对所述靶的推定序列进行折叠可以包括:使用定向邻接鉴定所述靶的推定序列的簇;以及使用鉴定的所述靶的推定序列的簇对所述测序数据进行折叠。在一些实施例中,对与所述靶的推定序列相关联的所述分子标记序列进行折叠包括:使用定向邻接对与所述靶的推定序列相关联的所述分子标记序列进行折叠。使用定向邻接对与所述靶的推定序列相关联的所述分子标记序列进行折叠可以包括:使用定向邻接鉴定与所述靶的推定序列相关联的所述分子标记序列的簇;以及使用鉴定的与所述靶的推定序列相关联的所述分子标记序列的簇对所述测序数据进行折叠。
在一些实施例中,所述方法包括:鉴定与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列,其中,与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列的出现小于不与所述靶的一个或多个嵌合序列相对应的所述靶的剩余一个或多个推定序列的出现;以及从所述测序数据中去除与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列。鉴定与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列可以包括:鉴定与所述多个分子序列中的一个分子标记序列相关联的所述靶的推定序列;鉴定与所述一个分子标记序列相关联的所述靶的推定序列中的推定序列,所述一个分子标记序列的出现小于与所述靶的一个或多个嵌合序列中的嵌合序列相对应的嵌合出现阈值。嵌合出现阈值的值可以是与一个分子标记序列相关联的所述靶的推定序列中的推定序列的出现,所述出现大于所述靶的推定序列中的任何其他序列的出现。
在一些实施例中,所述方法包括:确定所述测序数据中的所述靶的测序状态;以及确定与所述测序数据中的所述靶的推定序列相关联的噪音分子标记序列的出现,其中,估计的所述靶的出现与跟所述测序数据中的所述靶的推定序列相关联的所述分子标记序列的出现相关,所述测序数据根据所述噪音分子标记序列的出现进行了调整。所述测序数据中的所述靶的测序状态可以是饱和测序、测序不足、或过度测序。
在一些实施例中,所述测序不足状态可以是通过具有小于预定的测序不足阈值的深度的靶来确定,并且其中,所述靶的深度包括与所述测序数据中的所述靶的推定序列相关联的所述分子标记序列的平均、最小、或最大深度。测序不足阈值可以是约四。测序不足阈值可以独立于分子标记序列的数量。如果所述测序数据中的所述靶的测序状态是所述测序不足状态,则确定的噪音分子标记序列的数量可以是零。
在一些实施例中,所述饱和测序状态是通过与所述靶的推定序列相关联的所述分子标记序列的数量大于饱和阈值来确定。如果与所述靶的推定序列相关联的所述分子标记序列中的分子标记序列具有从大约6561个分子标记序列选择的序列,则所述饱和阈值可以是约6557。如果与所述靶的推定序列相关联的所述分子标记序列中的分子标记序列具有从大约65536个分子标记序列选择的序列,则所述预定的饱和阈值可以是约65532。如果所述测序数据中的所述靶的测序状态是所述饱和测序状态,则确定的噪音分子标记序列的数量可以是零。
在一些实施例中,所述过度测序状态是通过具有大于预定的过度测序阈值的深度的靶来确定,其中,所述靶的深度包括与所述测序数据中的所述靶的推定序列相关联的所述分子标记序列的平均、最小、或最大深度。如果与所述靶的推定序列相关联的所述分子标记序列中的分子标记序列具有从大约6561个分子标记序列选择的序列,则所述过度测序阈值可以是约250。在一些实施例中,所述方法可以包括:如果所述测序数据中的所述靶的测序状态是饱和测序状态或过度测序状态:将与所述测序数据中的所述靶的序列相关联的分子标记序列的数量进行二次取样至大约所述预定的过度测序阈值。
在一些实施例中,确定与所述测序数据中的所述靶的推定序列相关联的噪音分子标记序列的出现包括:如果负二项分布拟合条件是满足的,将信号负二项分布拟合至与所述测序数据中的所述靶的序列相关联的所述分子标记序列的出现,其中,所述信号负二项分布与跟所述测序数据中的所述靶的序列(作为信号分子标记序列)相关联的分子标记序列的出现相对应;将噪音负二项分布拟合至与所述测序数据中的所述靶的序列相关联的所述分子标记序列的出现,其中,所述噪音负二项分布与跟所述测序数据中的所述靶的序列(作为噪音分子标记序列)相关联的分子标记序列的出现相对应;以及使用所述信号负二项分布以及所述噪音负二项分布来确定所述噪音分子标记序列的出现。在一些实施例中,所述负二项分布拟合条件可以包括:所述测序数据中的所述靶的测序状态不是所述测序不足状态或所述过度测序状态。使用所述信号负二项分布以及所述噪音负二项分布来确定噪音分子标记序列的数量可以包括:针对与所述测序数据中的所述靶的推定序列相关联的所述分子标记序列中的每一个:确定所述分子标记序列在所述信号负二项分布中的信号概率;确定所述分子标记序列在所述噪音负二项分布中的噪音概率;以及如果所述信号概率小于所述噪音概率,将所述分子标记序列确定为噪音分子标记。
在一些实施例中,确定与所述测序数据中的所述靶的序列相关联的噪音分子标记序列的出现包括:如果所述测序数据中的所述靶的测序状态不是所述测序不足状态或所述过度测序状态并且与所述测序数据中的所述靶的序列相关联的分子标记序列的出现小于伪点阈值,则在确定与所述测序数据中的所述靶的序列相关联的噪音分子标记序列的出现之前,向与所述测序数据中的所述靶的序列相关联的分子标记序列的出现中添加伪点。所述伪点阈值可以是十。确定与所述测序数据中的所述靶的序列相关联的噪音分子标记序列的出现可以包括:如果所述测序数据中的所述靶的测序状态不是所述测序不足状态或所述过度测序状态并且与所述测序数据中的所述靶的序列相关联的分子标记序列的出现不小于伪点阈值,则在确定与所述测序数据中的所述靶的序列相关联的噪音分子标记序列的出现时去除非独特分子标记序列。
在一些实施例中,接收所述多个靶的测序数据包括:使用多个条形码(例如,随机条形码)使多个靶条形码化(例如,随机条形码化)以创建多个经条形码化的靶(例如,经随机条形码化的靶),其中,所述多个条形码中的每一个包括细胞标记和分子标记,其中,所述多个条形码中的至少两个条形码的分子标记包括不同的分子标记序列,并且其中所述多个条形码中的至少两个条形码包括具有相同的细胞标记序列的细胞标记;以及获得所述经条形码化的靶的测序数据。使用所述多个条形码使所述多个细胞中的所述多个靶条形码化(例如,随机条形码化)以为所述多个细胞中的细胞创建多个经条形码化的靶可以包括:使用颗粒的多个条形码使所述多个靶条形码化(例如,随机条形码化)以创建多个经条形码化的靶,其中,所述颗粒包括多个条形码的子集,其中,所述条形码的子集中的每一个包括相同的细胞标记序列并且具有至少100个不同的分子标记序列。
在一些实施例中,所述颗粒是珠。所述珠可以选自由以下项组成的分组:链霉抗生物素蛋白珠、琼脂糖珠、磁珠、缀合珠、蛋白a缀合珠、蛋白g缀合珠、蛋白a/g缀合珠、蛋白l缀合珠、寡聚(dt)缀合珠、二氧化硅珠、二氧化硅样珠、抗生物素微珠、抗荧光染料微珠、及其任何组合。所述颗粒可以包括选自由以下项组成的分组的材料:聚二甲基硅氧烷(pdms)、聚苯乙烯、玻璃、聚丙烯、琼脂糖、明胶、水凝胶、顺磁物质、陶瓷、塑料、玻璃、甲基苯乙烯、丙烯酸聚合物、钛、乳胶、琼脂糖凝胶、纤维素、尼龙、硅酮、及其任何组合。所述颗粒的条形码(例如,随机条形码)包括具有至少1000个、10000个或其任何组合的不同的分子标记序列的分子标记。
在一些实施例中,所述条形码(例如,随机条形码)的所述分子标记包括随机序列。所述颗粒可以包括至少10000个条形码。使用所述多个条形码(例如,随机条形码)使所述多个靶条形码化(例如,随机条形码化)以创建多个经条形码化的靶(例如,经随机条形码化的靶)可以包括:(i)使所述靶的拷贝与所述条形码的靶结合区接触;以及(ii)使用所述多个条形码逆转录所述多个靶以创建多个经逆转录的靶。在一些实施例中,所述方法包括:在获得所述多个经条形码化的靶的测序数据之前,扩增所述经条形码化的靶以生成多个经条形码化的靶(例如,经扩增的随机条形码化的靶)。扩增所述经条形码化的靶以生成多个经随机条形码化的靶可以包括:通过聚合酶链式反应(pcr)扩增所述经条形码化的靶。
在一些实施例中,披露了用于确定靶的出现的计算机系统。所述计算机系统可以包括:硬件处理器;以及具有存储在其上的指令的非暂态存储器,所述指令当被所述硬件处理器执行时使得所述处理器执行如以上权利要求中任一项所述的方法。披露了计算机可读介质。在一些实施例中,所述计算机可读介质包括用于执行如以上权利要求中任一项所述的方法的可执行代码。
附图说明
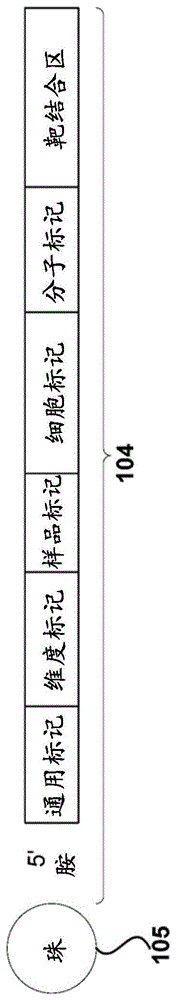
图1展示了非限制性示例性随机条形码。
图2示出了随机条形码化和数字计数的非限制性示例性工作流程。
图3是示出了用于从多个靶产生经随机条形码化的靶的索引文库的非限制性示例性过程的示意图。
图4是示出了分子标记错误、样品标记错误、和真分子标记信号的非限制性示例性分布的示意图。
图5是示出了基于定向邻接使用分子标记校正pcr和测序错误的非限制性示例性实施例的流程图。
图6是示出了基于递归取代错误校正和基于分布的错误校正来校正pcr和测序错误的非限制性示例性实施例的流程图。
图7是示出了基于递归取代错误校正的免疫受体条形码校正的非限制性示例性实施例的示意图。
图8是示出了校正核苷酸序列和分子标记中的错误(使用递归取代错误校正)以及校正归因于pcr嵌合体的测序数据中的错误的非限制性示例性实施例的流程图。
图9是免疫受体嵌合体的一种可能来源的示意图。
图10示出了适用于本披露的方法中使用的非限制性示例性仪器。
图11展示了可以结合本披露的实施例使用的计算机系统的非限制性示例性架构。
图12展示了示出具有适用于本披露的方法中使用的多个计算机系统的网络的非限制性示例性架构。
图13展示了根据本披露的方法使用共享的虚拟地址存储空间的多处理器计算机系统的非限制性示例性架构。
图14是用作输入分子增加的独特分子标记的理论计算的示例性图。
图15是示出了高表达基因-atcb的微孔板的每个分子标记的分子标记覆盖的示例性图,其中在错误分子标记和真实分子标记之间观察到不同的分布。
图16是示出了对于高表达基因-atcb的微孔板的每个分子标记的分子标记覆盖拟合两个负二项分布的示例性图。两个负二项分布的拟合证明具有较低的分子标记深度的分子标记错误和具有较高的分子标记深度的真分子标记可以在统计学上区分。x轴是分子深度。
图17示出了分子标记校正,其中为1的成对汉明距离被过度代表。在分子标记校正后,将相距一汉明距离的分子标记聚簇并折叠为相同的亲本分子标记。
图18示出了校正的分子标记数量相比于校正的读数覆盖数量的曲线。
图19示出了递归取代错误校正的实例的示意图。
图20a至20c示出了基于cd69的两个负二项分布校正pcr和测序错误的示例性结果。
图21a至21c示出了基于cd3e的两个负二项分布校正pcr和测序错误的示例性结果。
图22a至22j示出了使用两个负二项分布校正的数据集的非限制性示例性验证。
图23a至23d示出了来自96孔混合的jurkat和乳腺癌(brca)单细胞(检查的86个基因)的precisetm靶向测定的示例性t-随机邻域嵌入(t-sne)可视化。
图24a至24b是示出了在通过dbscan计算并通过每个簇中的基因标志物水平确定的两个选择的簇中具有>0个ml的基因的细胞簇之间的差异表达分析的非限制性示例性图。
图25a至25d是示出了来自96孔板的混合的jurkat和乳腺癌(t47d)单细胞(具有检查的86个基因)的bdprecisetm靶向测定的t-随机邻域嵌入(t-sne)可视化的非限制性示例性图。
图26a至26b是在任何错误校正步骤(图26a中示出的原始ml)之前、以及在rsec和dbec校正(图26b中示出的经调整的ml)之后,通过图25a至25d中鉴定的不同细胞簇之间的分子标记计数显示差别基因表达的非限制性示例性热图。
图27a至27b示出了展示使用递归取代错误校正的免疫受体条形码错误校正的非限制性实例的表。
图28是示出了免疫受体条形码错误校正的非限制性示例性结果的直方图。
具体实施方式
在以下详细说明中参考了形成本文的一部分的附图。在附图中,除非上下文另有指示,否则相似的符号通常标识相似的部件。在具体实施例、附图和权利要求中描述的说明性实施例不意味着是限制性的。在不脱离本文提出的主题的精神或范围的情况下,可以利用其他实施例,并且可以做出其他改变。容易理解的是,如本文一般描述的以及图中说明的本披露的方面能以各种不同的配置来布置、替换、组合、分离和设计,所有这些都在本文中明确考虑并且构成本披露内容的一部分。
来自genbank的所有专利、公开的专利申请、其他出版物、和序列,以及本文提及的其他数据库关于相关技术通过引用以其整体并入。
对少量核酸(例如信使核糖核苷酸(mrna)分子)进行量化对于确定例如在不同发育阶段或在不同环境条件下在细胞中表达的基因是临床上重要的。然而,确定核酸分子(例如,mrna分子)的绝对数量也是非常具有挑战性的,尤其是当分子数量非常小时。确定样品中分子的绝对数量的一种方法是数字聚合酶链式反应(pcr)。理想地,pcr在每个循环中产生分子的相同拷贝。然而,pcr可具有缺点使得每个分子复制具有随机概率,且此概率根据pcr循环和基因序列而变化,这导致扩增偏差和不准确的基因表达测量。可以将具有独特分子标记的随机条形码(也称为分子索引(mi)或通用分子索引(umi))用于对分子的数量进行计数并且校正扩增偏差。诸如precisetm测定(细胞研究公司(cellularresearch,inc.)(帕洛阿尔托,加利福尼亚))的随机条形码化可以通过使用分子标记(ml)在逆转录(rt)期间标记mrna来校正由pcr和文库制备步骤诱导的偏差。
precisetm测定可利用具有大量的(例如6561至65536个)随机条形码的非耗尽性池、聚(t)寡核苷酸上的独特分子标记,以在rt步骤期间与样品中的所有聚(a)-mrna杂交。除分子标记外,随机条形码的样品标签(也称为样品索引(si))可用于鉴定precisetm板的每个孔。随机条形码可包括通用pcr引发位点。在rt期间,靶基因分子与随机条形码任意地反应。每个靶分子可以与随机条形码杂交,从而产生随机地条形码化的互补核糖核苷酸(cdna)分子。在标记后,可将来自微孔板微孔的随机地条形码化的cdna分子池化进单个管中用于pcr扩增和测序。可以分析原始测序数据以产生读数的数量、具有独特分子标记的随机条形码的数量、以及基于泊松校正或基于两个负二项分布的校正方法的mrna分子的数量。
除了偏差校正之外,分子标记还可以通过揭示观察到的测序读数中存在的cdna分子的起始数量来更好地理解结果的统计质量。例如,大量读数可以指示统计学上准确的答案,但是如果读数仅来自少量起始mrna分子,则测量准确度可能会受到影响。
虽然通过pcr和文库制备步骤诱导的扩增偏差可以通过例如分子标记来纠正,但由于若干种其他因素,分子绝对数量的定量仍然是具有挑战性的。首先,mrna分子数量的估计可受到分子标记的总体多样性的限制。在随机条形码化中,mrna分子可以与可用的随机条形码任意地进行反应。因此,每个mrna分子可以与随机条形码杂交;然而,他的分子标记对于任何给定基因可能不一定是独特的。当mrna分子的数量相对于随机条形码的数量较小时,每个mrna分子可能与具有独特分子标记的随机条形码杂交,并且计数分子的数量可以等同于计数分子标记的数量。
随着mrna分子数量的增加,多个mrna分子变得越来越可能与具有相同分子标记的随机条形码杂交。因此,使用独特分子标记的计数可以低估了分子的数量。在一些情况下,可以基于泊松校正或基于总共观察到的独特分子标记的数量的两个负二项分布的校正来估计mrna分子的数量。然而,在观察到6561个随机条形码的整个集合的极端情况下,泊松校正或基于两个负二项分布的校正不再可能。例如,无论65000个或100000个起始mrna分子,在任何情况下预计最多6561个饱和随机条形码。
其次,pcr错误(即pcr扩增期间发生的错误)可以引入人工随机条形码并随意增加分子标记计数。第三,pcr扩增偏差和低效pcr可生成与错误无法区分的低拷贝的条形码分子。第四,测序错误(随机条形码序列的不准确调用)可以引入人工随机条形码并使分子标记计数增加。另外,测序深度可以是重要的,尤其是当测序太浅而不能检测样品文库中存在的所有随机条形码化的mrna时。
在执行免疫受体测序和分析时,可能会发生取代错误、引物交叉错误和pcr嵌合体错误。例如,当确定对免疫受体(诸如t细胞受体)进行编码的mrna分子的出现或拷贝数量时,可能发生这种错误。免疫受体是高度多样化的紧密相关的基因。因此,当与其他基因进行比较时,执行免疫受体测序和分析时此类错误的可能性可能会更高。所述错误通常导致免疫组库多样性的过高计量。减轻这些错误的方法在本文中被称为免疫受体条形码错误校正。在一些实施例中,免疫受体条形码错误校正利用递归取代错误校正来校正分子标记和核苷酸序列中的取代错误(例如,互补决定区3(cdr3)中的取代错误)。对于给定的样品标记或细胞标记,许多不同的cdr3可以与相同的分子标记序列相关联,从而导致对免疫受体多样性的过高估计。所述方法可以校正在进行分子标记和进行样品标记之前交叉的pcr嵌合体,然后通过基于分布的错误校正来鉴定和去除错误分子标记。
本文披露的内容包括用于确定靶的出现的方法。在一些实施例中,所述方法包括:(a)使用多个条形码(例如,随机条形码)使多个靶条形码化(例如,随机条形码化)以创建多个经条形码化的靶(例如,经随机条形码化的靶),其中,所述多个条形码中的每一个包括细胞标记和分子标记,其中,所述多个条形码中的至少两个条形码的分子标记包括不同的分子标记序列,并且其中所述多个条形码中的至少两个条形码包括具有相同的细胞标记序列的细胞标记;(b)获得所述经条形码化的靶的测序数据;以及(c)针对所述多个靶中的至少一个靶:(i)鉴定所述测序数据中的所述靶的推定序列;(ii)对与(i)中鉴定的所述测序数据中的所述靶的推定序列相关联的分子标记序列的出现进行计数;(iii)鉴定所述靶的推定序列的簇;(iv)使用(iii)中鉴定的所述靶的推定序列的簇对获得的测序数据进行折叠;(v)鉴定与所述靶的推定序列相关联的所述分子标记序列的簇;(vi)使用(v)中鉴定的所述分子标记序列的簇对所述测序数据进行折叠;(vii)鉴定组合序列的簇,其中,每个组合序列包括所述靶的序列中的序列以及所述分子标记序列中的相关联的分子标记序列;(viii)使用(vii)中鉴定的所述组合序列的簇对所述测序数据进行折叠;(ix)鉴定与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列,其中,与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列的出现小于不与所述靶的一个或多个嵌合序列相对应的所述靶的剩余一个或多个推定序列的出现;(x)从所述测序数据中去除(ix)中鉴定的与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列;以及(xi)估计所述靶的出现,其中,在(iv)、(vi)和(viii)对所述测序数据进行折叠并且在(x)中去除与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列后,估计的所述靶的出现与(ii)中计数的分子标记序列的数量相关。
本文披露的是用于确定靶的出现的方法。在一些实施例中,所述方法包括:(a)接收多个靶的测序数据,其中,所述测序数据包括所述多个靶中的靶的推定序列以及与所述测序数据中的所述靶的序列相关联的分子标记序列的出现;(b)对所述靶的推定序列进行折叠;(c)对与所述靶的推定序列相关联的分子标记序列进行折叠;以及(d)估计所述靶的出现,其中在(b)中对所述靶的推定序列的出现进行折叠并且对在(c)中确定的噪音分子标记序列的出现进行折叠之后,估计的所述靶的出现与跟所述测序数据中的所述靶的推定序列相关联的分子标记序列的出现相关。
披露了用于确定靶的出现的计算机系统。披露了包含可执行代码的非暂态计算机可读介质,所述代码在被执行时使得一个或多个计算装置确定靶的出现。
定义
除非另外定义,本文所用的技术术语和科学术语具有与本披露所属领域的普通技术人员通常所理解的相同意义。参见,例如,singleton等人,dictionaryofmicrobiologyandmolecularbiology[微生物学和分子生物学词典],第2版,约翰·威利父子出版公司(j.wiley&sons),(纽约,纽约州1994);sambrook等人,分子克隆实验室手册(molecularcloning,alaboratorymanual),冷泉港出版社(coldspringsharborpress)(纽约冷泉港(coldspringsharbor,ny)1989)。出于本披露的目的,以下术语定义如下。
如本文所用的,术语“衔接子”可以意指促进相关联的核酸的扩增或测序的序列。相关联的核酸可包括靶核酸。相关联的核酸可以包括空间标记、靶标记、样品标记、索引标记、或条形码序列(例如,分子标记)中的一个或多个。衔接子可以是线性的。衔接子可以是预腺苷酸化的衔接子。衔接子可以是双链或单链的。一个或多个衔接子可以位于核酸的5’或3’端。当衔接子在5’和3’端包括已知序列时,已知序列可以是相同或不同的序列。位于多核苷酸的5’和/或3’端的衔接子能够与固定在表面上的一个或多个寡核苷酸杂交。在一些实施例中,衔接子可以包括通用序列。通用序列可以是两个或更多个核酸分子共有的核苷酸序列的区域。两个或更多个核酸分子也可具有不同序列的区域。因此,例如,5’衔接子可以包括相同和/或通用核酸序列,并且3’衔接子可以包括相同和/或通用序列。可存在于多个核酸分子的不同成员中的通用序列可允许使用与通用序列互补的单个通用引物复制或扩增多个不同序列。相似地,可以存在于核酸分子的集合中的不同成员中的至少一个、两个(例如,一对)或更多个通用序列可以允许使用与通用序列互补的至少一个、两个(例如,一对)或更多个单个通用引物复制或扩增多个不同序列。因此,通用引物包括可与此类通用序列杂交的序列。可以修饰携带靶核酸序列的分子以将通用衔接子(例如,非靶核酸序列)附接至不同靶核酸序列的一端或两端。与靶核酸附接的一个或多个通用引物可以提供通用引物杂交的位点。与靶核酸附接的一个或多个通用引物可以彼此相同或不同。
如本文使用的,术语“关联”或“与……相关联”可意指两个或更多个种类可以被鉴定为在某个时间点处共定位。关联可意指两个或更多个种类在或曾经在相似的容器内。关联可以是信息学关联。例如,关于两个或更多个种类的数字信息可以被存储并且可以用于确定所述种类中的一个或多个在某个时间点处共定位。关联也可以是物理关联。在一些实施例中,两个或更多个相关联的种类彼此之间或与共同的固体或半固体表面是“连接的”、“附接的”或“固定的”。关联可以指用于将标记附接到固体或半固体支持物(例如珠)上的共价或非共价方式。关联可以是靶与标记之间的共价键。关联可以包括两个分子(例如靶分子和标记)之间的杂交。
如本文使用的,术语“互补性”可以指两个核苷酸之间精确配对的能力。例如,如果核酸的在给定位置的核苷酸能够与另一个核酸的核苷酸以氢键结合,则两个核酸被认为在所述位置处是彼此互补的。两单链核酸分子之间的互补性可以是“部分的”,其中所述核苷酸中仅一些结合,或者当所述单链分子之间存在完全互补性时,这种互补性可以是完全的。如果第一核苷酸序列与第二核苷酸序列互补,则可以认为第一核苷酸序列是第二序列的“互补体”。如果第一核苷酸序列互补于和第二序列相反的序列(即,核苷酸顺序相反),则可以认为第一核苷酸序列是第二序列的“反向互补体”。如本文使用的,术语“互补体”、“互补”和“反向互补体”可以互换使用。从本披露可以理解,如果一个分子可以与另一个分子杂交,则其可以是杂交的分子的互补体。
如本文使用的,术语“数字计数”可以指用于估计样品中靶分子数量的方法。数字计数可以包括确定已经与样品中的靶相关联的独特标记的数量的步骤。这种在性质上可以随机的方法将计数分子的问题从相同分子的定位和鉴定之一转化为有关检测一组预定义标记的一系列是/否数字问题。
如本文使用的,术语(一个或多个)“标记”可以指与样品中的靶相关联的核酸代码。标记可以是例如核酸标记。标记可以是完全或部分可扩增的标记。标记可以是完全或部分可测序的标记。标记可以是可鉴定为有区别的天然核酸的一部分。标记可以是已知的序列。标记可以包括核酸序列的接点,例如天然和非天然序列的接点。如本文使用的,术语“标记”可以与术语“索引”、“标签”或“标记-标签”互换使用。标记可以传达信息。例如,在各种实施例中,可以使用标记来确定样品的身份、样品的来源、细胞的身份和/或靶。
如本文使用的,术语“非耗尽性储库(non-depletingreservoir)”可以指由许多不同标记组成的条形码(例如,随机条形码)的池。非耗尽性储库可以包括大量不同的条形码,使得当非耗尽性储库与靶池相关联时,每个靶可能与独特的条形码相关联。每个经标记的靶分子的独特性可以通过随机选择的统计来确定,并且取决于与多样的标记相比在集合中相同的靶分子的拷贝数。所得的经标记的靶分子集合的大小可以通过条形码化处理的随机性质来确定,并且然后对检测到的条形码的数量的分析允许计算原始集合或样品中存在的靶分子的数量。当存在的靶分子的拷贝数量与独特的条形码的数量的比率低时,经标记的靶分子是高度独特的(即,用给定的标记来标记多于一个靶分子的概率非常低)。
如本文所用的,术语“核酸”是指多核苷酸序列、或其片段。核酸可以包括核苷酸。核酸对于细胞可以是外源的或内源的。核酸可以存在于无细胞环境中。核酸可以是基因或其片段。核酸可以是dna。核酸可以是rna。核酸可以包括一种或多种类似物(例如改变的骨架、糖或核碱基)。类似物的一些非限制性实例包括:5-溴尿嘧啶、肽核酸、外来核酸、吗啉代、锁核酸、二醇核酸、苏糖核酸、二脱氧核苷酸、虫草菌素、7-脱氮-gtp、荧光团(例如,罗丹明或与糖连接的荧光黄素)、含有核苷酸的硫醇、生物素连接的核苷酸、荧光基类似物、cpg岛、甲基-7-鸟苷、甲基化的核苷酸、肌苷、硫代尿苷、假尿苷、二氢尿苷、辫苷、以及怀俄苷。“核酸”、“多核苷酸”、“靶多核苷酸”和“靶核酸”可以互换使用。
核酸可以包括一种或多种修饰(例如,碱基修饰、骨架修饰),以为核酸提供新的或增强的特征(例如,改进的稳定性)。核酸可以包括核酸亲和标签。核苷可以是碱基-糖组合。核苷的碱基部分可以是杂环碱基。此类杂环碱基的两个最常见的类别是嘌呤和嘧啶。核苷酸可以是进一步包括与核苷的糖部分共价连接的磷酸基团的核苷。对于包括呋喃戊糖的那些核苷,磷酸基团可以连接到糖的2’、3’或5’羟基部分。在形成核酸中,磷酸基团可以将相邻的核苷彼此共价连接以形成线性高分子化合物。转而此线性高分子化合物的各自端可以进一步接合而形成环状化合物;然而,线性化合物通常是合适的。此外,线性化合物可以具有内部核苷酸碱基互补性,并且因此可以按产生完全或部分双链化合物的方式折叠。在核酸中,所述磷酸基团通常可以被称为形成核酸的核苷间骨架。连键或骨架可以是3’到5’磷酸二酯键。
核酸可以包括修饰的骨架和/或修饰的核苷间键。核酸可以包括由短链烷基或环烷基核苷间键、混合杂原子、和烷基或环烷基核苷间键或者一个或多个短链杂原子的或杂环的核苷间键形成的多核苷酸骨架。核酸可以包括核酸模拟物。核酸可以包括吗啉代骨架结构。核酸可以包括具有附接到吗啉代环上的杂环碱基的连接的吗啉代单元(即吗啉代核酸)。
核酸还可以包括核碱基(通常简称为“碱基”)修饰或取代。如本文使用的,“未修饰的”或“天然的”核碱基可以包括嘌呤碱基(例如腺嘌呤(a)和鸟嘌呤(g))、以及嘧啶碱基(例如胸腺嘧啶(t)、胞嘧啶(c)和尿嘧啶(u))。经修饰的核碱基可以包括其他合成以及天然的核碱基。
如本文使用的,术语“样品”可以指包括靶的组合物。用于通过披露的方法、装置、和系统进行分析的合适样品包括细胞、组织、器官、或生物体。
如本文使用的,术语“采样装置”或“装置”可以指可以取一部分样品和/或将所述部分放置在基底上的装置。采样装置可以指例如荧光激活细胞分选(facs)机、细胞分选机、活检针、活检装置、组织切片装置、微流体装置、叶栅和/或超薄切片机。
如本文使用的,术语“固体支持物”可以指可以附接多个条形码(例如,随机条形码)的离散固体或半固体表面。固体支持物可以包括任何类型的实心的、多孔的或空心的球体、球、承座、圆柱体或其他类似配置,其由塑料、陶瓷、金属或高分子材料(例如,水凝胶)构成,其上可以固定核酸(例如,共价地或非共价地)。固体支持物可以包括可以是球形的(例如,微球)或具有非球形或不规则形状的离散颗粒,所述形状是诸如立方形、长方形、锥形、圆柱形、圆锥形、椭圆形或圆盘形等。珠的形状可以是非球形的。以阵列间隔开的多个固体支持物可以不包括基底。固体支持物可以与术语“珠”互换使用。
固体支持物可以指“基底”。基底可以是一种固体支持物。基底可以指可以在其上进行本披露的方法的连续的固体或半固体表面。例如,基底可以指阵列、盒、芯片、装置和载玻片。
如本文使用的,术语“空间标记”可以指其可以在空间中与位置相关联的标记。
如本文使用的,术语“随机条形码”可以指包括本披露的标记的多核苷酸序列。随机条形码可以是可用于随机条形码化的多核苷酸序列。随机条形码可用于对样品中的靶定量。随机条形码可用于控制标记与靶相关联后可能发生的错误。例如,随机条形码可用于评估扩增或测序错误。与靶相关联的随机条形码可以称为随机条形码-靶或随机条形码-标签-靶。
如本文所用,术语“基因特异性随机条形码”可以指包括标记和基因特异性的靶结合区的多核苷酸序列。随机条形码可以是可用于随机条形码化的多核苷酸序列。随机条形码可用于对样品中的靶定量。随机条形码可用于控制标记与靶相关联后可能发生的错误。例如,随机条形码可用于评估扩增或测序错误。与靶相关联的随机条形码可以称为随机条形码-靶或随机条形码-标签-靶。
如本文使用的,术语“随机条形码化”可以指核酸的随机标记(例如,条形码化)。随机条形码化可以利用递归泊松策略来关联并对与靶相关联的标记进行定量。如本文使用的,术语“随机条形码化”可以与“随机进行标记”互换使用。
如本文使用的,术语“靶”可以指可与条形码(例如,随机条形码)相关联的组合物。用于通过披露的方法、装置和系统进行分析的示例性合适的靶包括寡核苷酸、dna、rna、mrna、微小rna、trna等。靶可以是单链的或双链的。在一些实施例中,靶可以是蛋白质、肽或多肽。在一些实施例中,靶是脂质。如本文使用的,“靶”可以与“种类”互换使用。
如本文所用的,术语“逆转录酶”可以指具有逆转录酶活性(即,催化从rna模板合成dna)的一组酶。通常,这样的酶包括但不限于逆转录病毒逆转录酶、逆转录转座子逆转录酶、逆转录质粒逆转录酶、逆转录子逆转录酶、细菌逆转录酶、ii型内含子衍生的逆转录酶,及其突变体、变体或衍生物。非逆转录病毒逆转录酶包括非ltr逆转录转座子逆转录酶、逆转录质粒逆转录酶、逆转录子逆转录酶和ii型内含子逆转录酶。ii型内含子逆转录酶的实例包括乳酸乳球菌li.ltrb内含子逆转录酶、细长嗜热聚球藻(thermosynechococcuselongatus)tei4c内含子逆转录酶或嗜热脂肪土芽孢杆菌gsi-iic内含子逆转录酶。其他类别的逆转录酶可以包括许多类型的非逆转录病毒逆转录酶(即,逆转录子、ii型内含子、以及多样性产生型逆转录元件等等)。
术语“通用衔接子引物”、“通用引物衔接子”或“通用衔接子序列”可互换地使用是指可以用于与条形码(例如,随机条形码)杂交以产生基因特异性条形码的核苷酸序列。通用衔接子序列例如可以是在本披露的方法中使用的所有条形码通用的已知的序列。例如,当使用本文披露的方法标记多个靶时,每个靶特异性序列可以连接到相同的通用衔接子序列上。在一些实施例中,超过一个通用衔接子序列可以用于本文披露的方法中。例如,当使用本文披露的方法标记多个靶时,至少两个靶特异性序列连接到不同的通用衔接子序列上。通用衔接子引物及其互补物可以包括在两个寡核苷酸中,其中的一个包括靶特异性序列且另一个包括条形码。例如,通用衔接子序列可以是包括靶特异性序列的寡核苷酸的一部分,以产生与靶核酸互补的核苷酸序列。包括条形码和通用衔接子序列的互补序列的第二寡核苷酸可与核苷酸序列杂交并产生靶特异性条形码(例如,靶特异性随机条形码)。在一些实施例中,通用衔接子引物具有与本披露内容的方法中使用的通用pcr引物不同的序列。
本文披露的是用于检测和/或校正在pcr和/或测序期间发生的错误的方法和系统。错误的类型可以变化,例如,包括但不限于取代错误(一个或多个碱基)和非取代错误。在取代错误中,一个碱基取代错误的发生频率远高于一个碱基不同的那些。可以使用所述方法和系统例如以通过随机条形码化提供分子靶的精确计数。
条形码
条形码化(诸如随机条形码化)已描述于例如us20150299784、wo2015031691、以及fu等人,procnatlacadsciu.s.a.[美国国家科学院院刊]2011年5月31日;108(22):9026-31中,这些出版物的内容以其整体结合在此。在一些实施例中,本文披露的条形码可以是随机条形码,所述随机条形码可以是可用于对靶进行随机标记(例如,条形码,标签)的多核苷酸序列。如果随机条形码的不同的条形码序列的数量与待标记的任何靶的出现的数量的比率可以是、或约1:1、2:1、3:1、4:1、5:1、6:1、7:1、8:1、9:1、10:1、11:1、12:1、13:1、14:1、15:1、16:1、17:1、18:1、19:1、20:1、30:1、40:1、50:1、60:1、70:1、80:1、90:1、100:1、或在这些值的任何两个之间的数字或范围,则条形码可以称为随机条形码。靶可以是包括具有相同或几乎相同序列的mrna分子的mrna种类。如果随机条形码的不同的条形码序列的数量与待标记的任何靶的出现的数量的比率是至少、或至多1:1、2:1、3:1、4:1、5:1、6:1、7:1、8:1、9:1、10:1、11:1、12:1、13:1、14:1、15:1、16:1、17:1、18:1、19:1、20:1、30:1、40:1、50:1、60:1、70:1、80:1、90:1、或100:1,则条形码可以称为随机条形码。随机条形码的条形码序列可以称为分子标记。
条形码(例如随机条形码)可以包括一个或多个标记。示例性标记可以包括通用标记、细胞标记、条形码序列(例如,分子标记)、样品标记、板标记、空间标记、和/或前空间(pre-spatial)标记。图1展示了具有空间标记的示例性条形码104。条形码104可以包括可将条形码与固体支持物105连接的5’胺。条形码可以包括通用标记、维度标记、空间标记、细胞标记、和/或分子标记。条形码中不同标记(包括但不限于通用标记、维度标记、空间标记、细胞标记、和分子标记)的顺序可以改变。例如,如图1中显示,通用标记可以是5’-末端标记,且分子标记可以是3’-末端标记。空间标记、维度标记、和细胞标记可以处于任何顺序。在一些实施例中,通用标记、空间标记、维度标记、细胞标记、和分子标记是处于任何顺序的。条形码可以包括靶结合区。靶结合区可以与样品中的靶(例如,靶核酸、rna、mrna、dna)相互作用。例如,靶结合区可以包括可以与mrna的聚(a)尾相互作用的寡聚(dt)序列。在一些情况下,条形码的标记(例如,通用标记、维度标记、空间标记、细胞标记和条形码序列)可以由1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、或20个或更多个核苷酸分开。
标记(例如,细胞标记)可以包括一组独特的定义长度的核酸子序列,例如每个七个核苷酸(相当于一些汉明错误校正代码中使用的比特数量),其可以设计为提供错误校正能力。可以设计包括七个核苷酸序列的错误校正子序列组,使得所述组中的序列的任何成对组合展现出定义的“遗传距离”(或错配碱基数),例如一组纠错子序列能被设计为展现三个核苷酸的遗传距离。在这种情况下,对于经标记的靶核酸分子的序列数据组中的错误校正序列的审查(在下面更全面地描述)能允许检测或校正扩增或测序错误。在一些实施例中,用于产生错误校正代码的核酸子序列的长度可以变化,例如,它们可以是、或约1、2、3、4、5、6、7、8、9、10、15、20、30、31、40、50个、或在这些值的任何两个之间的数字或范围的核苷酸长度。在一些实施例中,其他长度的核酸子序列可以用来产生错误校正代码。
条形码(例如,随机条形码)可以包括靶结合区。靶结合区可以与样品中的靶相互作用。所述靶可以是、或包括核糖核酸(rna)、信使rna(mrna)、微小rna、小干扰rna(sirna)、rna降解产物、各自含有聚(a)尾的rna、或其任何组合。在一些实施例中,多个靶可以包括脱氧核糖核酸(dna)。
在一些实施例中,靶结合区可以包括可以与mrna的聚(a)尾相互作用的寡聚(dt)序列。条形码的一个或多个标记(例如,通用标记、维度标记、空间标记、细胞标记、和条形码序列(例如,分子标记))可以通过间隔物与条形码的剩余标记的另一个或两个分开。间隔物可以是例如,1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、或20个或更多个核苷酸。在一些实施例中,条形码的标记中没有标记被间隔物分开。
通用标记
条形码可以包括一个或多个通用标记。在一些实施例中,对于条形码组中的所有条形码(附接到给定的固体支持物上的),一个或多个通用标记可以是相同的。在一些实施例中,对于附接到多个珠上的所有条形码,一个或多个通用标记可以是相同的。在一些实施例中,通用标记可以包括能够与测序引物杂交的核酸序列。测序引物可以用于对包括通用标记的条形码进行测序。测序引物(例如,通用测序引物)可以包括与高通量测序平台相联系的测序引物。在一些实施例中,通用标记可以包括能够与pcr引物杂交的核酸序列。在一些实施例中,通用标记可以包括能够与测序引物和pcr引物杂交的核酸序列。能够与测序或pcr引物杂交的通用标记的核酸序列可以被称为引物结合位点。通用标记可以包括可用于引发条形码转录的序列。通用标记可以包括可用于延伸条形码或条形码内的区域的序列。通用标记的长度可以是、或约1、2、3、4、5、10、15、20、25、30、35、40、45、50个核苷酸、或在这些值的任何两个之间的数字或范围的核苷酸。例如,通用标记可以包括至少约10个核苷酸。通用标记的长度可以是至少、或至多1、2、3、4、5、10、15、20、25、30、35、40、45、50、100、200、或300个核苷酸。在一些实施例中,可切割接头或修饰的核苷酸可以是通用标记序列的一部分,以使条形码能够从支持物上被切割下来。
维度标记
条形码可以包括一个或多个维度标记。在一些实施例中,维度标记可以包括提供关于标记(例如,随机标记)发生的维度的信息的核酸序列。例如,维度标记可以提供关于对靶进行条形码化的时间的信息。维度标记可以与样品中条形码化(例如,随机条形码化)的时间相关联。维度标记可以在标记的时间处被激活。不同的维度标记可以在不同的时间被激活。所述维度标记提供关于靶、靶组和/或样品被条形码化的顺序的信息。例如,在细胞周期的g0期可以对细胞群进行条形码化。在细胞周期的g1期,可以用条形码(例如,随机条形码)对所述细胞再次进行脉冲处理。在细胞周期的s期,可以用条形码对所述细胞再次进行脉冲处理,等等。每个脉冲(例如,细胞周期的每个阶段)处的条形码可以包括不同的维度标记。以这种方式,所述维度标记提供关于哪些靶在细胞周期的哪个时期被标记的信息。维度标记可以探询许多不同的生物阶段。示例性的生物学时间可以包括但不限于细胞周期、转录(例如,转录起始)和转录物降解。在另一个实例中,样品(例如,细胞、细胞群)可以在用药物和/或疗法治疗之前和/或之后标记。不同靶的拷贝数的变化可以指示样品对药物和/或疗法的反应。
维度标记可以是可激活的。可以在特定时间点激活可激活的维度标记。可激活的标记可以被例如组成性地激活(例如,不关闭)。所述可激活的维度标记可以被例如可逆地激活(例如,所述可激活的维度标记可以打开和关闭)。所述维度标记可以被例如可逆地激活至少1、2、3、4、5、6、7、8、9、10次或更多次。所述维度标记可以被可逆地激活例如至少1、2、3、4、5、6、7、8、9、10次或更多次。在一些实施例中,可以用荧光、光、化学事件(例如,切割,另一种分子的连接,修饰的添加(例如,聚乙二醇化、sumo化、乙酰化、甲基化、去乙酰化、去甲基化)、光化学事件(例如,光锁定)以及引入非天然的核苷酸将所述维度标记激活。
在一些实施例中,所述维度标记对于附接到给定的固体支持物(例如,珠)上的所有条形码(例如,随机条形码)可以是相同的,但对于不同的固体支持物(例如,珠)是不同的。在一些实施例中,相同固体支持物上的至少60%、70%、80%、85%、90%、95%、97%、99%或100%的条形码可以包括相同的维度标记。在一些实施例中,相同固体支持物上的至少60%的条形码可以包括相同的维度标记。在一些实施例中,相同固体支持物上的至少95%的条形码可以包括相同的维度标记。
多个固体支持物(例如,珠)可以表现多达106个或更多个独特维度标记序列。维度标记的长度可以是、或约1、2、3、4、5、10、15、20、25、30、35、40、45、50个核苷酸、或在这些值的任何两个之间的数字或范围的核苷酸。维度标记的长度可以是至少、或至多1、2、3、4、5、10、15、20、25、30、35、40、45、50、100、200、或300个核苷酸。维度标记可包括在约5至约200个之间的核苷酸。维度标记可以包括在约10至约150个之间的核苷酸。维度标记可以包括长度在约20至约125个之间的核苷酸。
空间标记
条形码可以包括一个或多个空间标记。在一些实施例中,空间标记可以包括提供与条形码相关联的靶分子的空间取向的信息的核酸序列。空间标记可以与样品中的坐标相关联。所述坐标可以是固定的坐标。例如,可以参考基底固定坐标。空间标记可以参考二维或三维网格。可以参考界标固定坐标。在空间中界标是可被鉴定的。界标可以是可被成像的结构。界标可以是生物学结构,例如解剖学界标。界标可以是细胞界标,例如细胞器。界标可以是非天然界标,诸如具有可鉴定标识(诸如色码、条形码、磁性、荧光、放射性或独特尺寸或形状)的结构。空间标记可以与物理分区(例如,孔、容器或液滴)相关联。在一些实施例中,将多个空间标记一起用于编码在空间中的一个或多个位置。
所述空间标记对于附接到给定的固体支持物(例如,珠)上的所有条形码可以是相同的,但对于不同的固体支持物(例如,珠)是不同的。在一些实施例中,包括相同空间标记的相同固体支持物上的条形码的百分比可以是、或约60%、70%、80%、85%、90%、95%、97%、99%、100%、或在这些值的任何两个之间的数字或范围。在一些实施例中,包括相同空间标记的相同固体支持物上的条形码的百分比可以是至少、或至多60%、70%、80%、85%、90%、95%、97%、99%、或100%。在一些实施例中,相同固体支持物上的至少60%的条形码可以包括相同的空间标记。在一些实施例中,相同固体支持物上的至少95%的条形码可以包括相同的空间标记。
多个固体支持物(例如,珠)可以表现多达106个或更多个独特空间标记序列。空间标记的长度可以是、或约1、2、3、4、5、10、15、20、25、30、35、40、45、50个核苷酸、或在这些值的任何两个之间的数字或范围的核苷酸。空间标记的长度可以是至少、或至多1、2、3、4、5、10、15、20、25、30、35、40、45、50、100、200、或300个核苷酸。空间标记可以包括在约5至约200个之间的核苷酸。空间标记可以包括在约10至约150个之间的核苷酸。空间标记可以包括长度在约20至约125个之间的核苷酸。
细胞标记
条形码(例如,随机条形码)可以包括一个或多个细胞标记。在一些实施例中,细胞标记可以包括提供用于确定哪个靶核酸来自哪个细胞的信息的核酸序列。在一些实施例中,所述细胞标记对于附接到给定的固体支持物(例如,珠)上的所有条形码是相同的,但对于不同的固体支持物(例如,珠)是不同的。在一些实施例中,包括相同细胞标记的相同固体支持物上的条形码的百分比可以是、或约60%、70%、80%、85%、90%、95%、97%、99%、100%、或在这些值的任何两个之间的数字或范围。在一些实施例中,包括相同细胞标记的相同固体支持物上的条形码的百分比可以是、或约60%、70%、80%、85%、90%、95%、97%、99%、或100%。例如,相同固体支持物上的至少60%的条形码可以包括相同的细胞标记。作为另一个实例,相同固体支持物上的至少95%的条形码可以包括相同的细胞标记。
多个固体支持物(例如,珠)可以表现多达106个或更多个独特细胞标记序列。细胞标记的长度可以是、或约1、2、3、4、5、10、15、20、25、30、35、40、45、50个核苷酸、或在这些值的任何两个之间的数字或范围的核苷酸。细胞标记的长度可以是至少、或至多1、2、3、4、5、10、15、20、25、30、35、40、45、50、100、200、或300个核苷酸。例如,细胞标记可以包括约5至约200个之间的核苷酸。作为另一个实例,细胞标记可以包括约10至约150个之间的核苷酸。还作为另一个实例,细胞标记可以包括长度在约20至约125个之间的核苷酸。
条形码序列
条形码可以包括一个或多个条形码序列。在一些实施例中,条形码序列可以包括为与条形码杂交的特定类型的靶核酸种类提供鉴定信息的核酸序列。条形码序列可以包括如下核酸序列,所述核酸序列为与条形码(例如,靶结合区)杂交的靶核酸种类的特定出现提供计数器(例如,提供粗略近似)。
在一些实施例中,将一组不同的条形码序列附接到给定的固体支持物(例如,珠)上。在一些实施例中,可以有、或约有102、103、104、105、106、107、108、109个、或在这些值的任何两个之间的数字或范围的独特分子标记序列。例如,多个条形码可以包括具有不同序列的约6561个条形码序列。作为另一个实例,多个条形码可以包括具有不同序列的约65536个条形码序列。在一些实施例中,可以有至少、或至多102、103、104、105、106、107、108、或109个独特条形码序列。独特分子标记序列可以附接到给定的固体支持物(例如,珠)上。
在不同实施方式中,条形码的长度可以是不同的。例如,条形码的长度可以是、或约1、2、3、4、5、10、15、20、25、30、35、40、45、50个、或在这些值的任何两个之间的数字或范围的核苷酸。作为另一个实例,条形码的长度可以是至少、或至多1、2、3、4、5、10、15、20、25、30、35、40、45、50、100、200、或300个核苷酸。
分子标记
条形码(例如,随机条形码)可以包括一个或多个分子标记。分子标记可以包括条形码序列。在一些实施例中,分子标记可以包括为与条形码杂交的特定类型的靶核酸种类提供鉴定信息的核酸序列。分子标记可以包括如下核酸序列,所述核酸序列为与条形码(例如,靶结合区)杂交的靶核酸种类的特定出现提供计数器。
在一些实施例中,将一组不同的分子标记附接到给定的固体支持物(例如,珠)上。在一些实施例中,可以有、或约有102、103、104、105、106、107、108、109个、或在这些值的任何两个之间的数字或范围的独特分子标记序列。例如,多个条形码可以包括具有不同序列的约6561个分子标记。作为另一个实例,多个条形码可以包括具有不同序列的约65536个分子标记。在一些实施例中,可以有至少、或至多102、103、104、105、106、107、108、或109个独特分子标记序列。具有独特分子标记序列的条形码可以附接到给定的固体支持物(例如,珠)上。
对于使用多个随机条形码的随机条形码化,不同分子标记序列的数量与任何靶的出现的数量的比率可以是、或约1:1、2:1、3:1、4:1、5:1、6:1、7:1、8:1、9:1、10:1、11:1、12:1、13:1、14:1、15:1、16:1、17:1、18:1、19:1、20:1、30:1、40:1、50:1、60:1、70:1、80:1、90:1、100:1、或在这些值的任何两个之间的数字或范围。靶可以是包括具有相同或几乎相同序列的mrna分子的mrna种类。在一些实施例中,不同分子标记序列的数量与任何靶的出现的数量的比率是至少、或至多1:1、2:1、3:1、4:1、5:1、6:1、7:1、8:1、9:1、10:1、11:1、12:1、13:1、14:1、15:1、16:1、17:1、18:1、19:1、20:1、30:1、40:1、50:1、60:1、70:1、80:1、90:1、或100:1。
分子标记的长度可以是或是约1、2、3、4、5、10、15、20、25、30、35、40、45、50个、或在这些值的任何两个之间的数字或范围的核苷酸。分子标记的长度可以是至少、或至多1、2、3、4、5、10、15、20、25、30、35、40、45、50、100、200、或300个核苷酸。
靶结合区
条形码可以包括一个或多个靶结合区,诸如捕获探针。在一些实施例中,靶结合区可以与目的靶杂交。在一些实施例中,所述靶结合区可以包括与靶(例如,靶核酸、靶分子,例如待分析的细胞核酸)进行特异性杂交(例如,与特定基因序列杂交)的核酸序列。在一些实施例中,靶结合区可以包括可附接(例如,杂交)至特定靶核酸的特定位置的核酸序列。在一些实施例中,靶结合区可以包括能够与限制性酶位点突出端(例如,ecori粘性末端突出端)进行特异性杂交的核酸序列。然后条形码可以连接到包括与限制性位点突出端互补的序列的任何核酸分子。
在一些实施例中,靶结合区可以包括非特异性靶核酸序列。非特异性靶核酸序列可以指独立于靶核酸的特定序列可与多个靶核酸结合的序列。例如,靶结合区可以包括与mrna分子上的聚(a)尾杂交的随机多聚体序列或寡聚(dt)序列。随机多聚体序列可以是例如随机二聚体、三聚体、四聚体、五聚体、六聚体、七聚体、八聚体、九聚体、十聚体或任何长度的更高多聚体序列。在一些实施例中,对于附接到给定珠的所有条形码,所述靶结合区是相同的。在一些实施例中,对于附接到给定珠上的多个条形码,靶结合区可以包括两个或更多个不同的靶结合序列。靶结合区的长度可以是或是约5、10、15、20、25、30、35、40、45、50个核苷酸、或在这些值的任何两个之间的数字或范围的核苷酸。靶结合区的长度可以是至多约5、10、15、20、25、30、35、40、45、50个或更多个核苷酸。
在一些实施例中,靶结合区可以包括寡聚(dt),所述寡聚(dt)可以与包括聚腺苷酸化末端的mrna杂交。靶结合区可以是基因特异性的。例如,可以将靶结合区配置为与靶的特定区域杂交。靶结合区的长度可以是或是约1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30个核苷酸、或在这些值的任何两个之间的数字或范围的核苷酸。靶结合区的长度可以是至少、或至多1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、或30个核苷酸。靶结合区的长度可以是约5至30个核苷酸。当条形码包括基因特异性靶结合区时,所述条形码可以称为基因特异性条形码。
定向特性
随机条形码(例如,随机条形码)可以包括一种或多种可用于定向(例如,比对)条形码的定向特性。条形码可以包括用于等电聚焦的部分。不同的条形码可以包括不同的等电聚焦点。当将这些条形码被引入样品中时,所述样品可以经历等电聚焦,以便于将所述条形码定位成已知的方式。以这种方式,所述定向特性可以用于开发样品中条形码的已知的映射。示例性定向特性可以包括电泳迁移率(例如,基于条形码的尺寸)、等电点、自旋、电导率和/或自组装。例如,条形码具有自组装的定向特性,当激活时可以自组装成特定定向(例如,核酸纳米结构)。
亲和特性
条形码(例如,随机条形码)可以包括一种或多种亲和特性。例如,空间标记可以包括亲和特性。亲和特性可以包括化学和/或生物部分,所述特性可以促进所述条形码与另一种实体(例如,细胞受体)的结合。例如,亲和特性可以包括抗体,例如,对于样品上的具体部分(例如,受体)特异性的抗体。在一些实施例中,抗体可以将条形码引导到特定细胞类型或分子上。在特定细胞类型或分子处的和/或附近的靶可以被标记(例如,随机地标记)。在一些实施例中,除了空间标记的核苷酸序列,亲和特性可以提供空间信息,因为所述抗体可以将所述条形码引导至特定位置。抗体可以是治疗性抗体,例如单克隆抗体或多克隆抗体。抗体可以是人源化的或嵌合的。抗体可以是裸抗体或融合抗体。
抗体可以是全长(即,天然存在的或通过正常免疫球蛋白基因片段重组过程形成的)免疫球蛋白分子(例如,igg抗体)或免疫球蛋白分子的免疫活性(即,特异性结合)部分(像抗体片段)。
抗体片段可以是例如抗体的一部分,诸如f(ab’)2、fab’、fab、fv、sfv等。在一些实施例中,抗体片段可以与由全长抗体识别的相同的抗原结合。抗体片段可以包括由抗体的可变区组成的分离的片段,诸如由重链和轻链的可变区组成的“fv”片段和其中轻链和重链可变区通过肽接头连接的重组单链多肽分子(“scfv蛋白”)。示例性抗体可以包括但不限于癌细胞抗体、病毒抗体、结合至细胞表面受体(cd8、cd34、cd45)的抗体、和治疗性抗体。
通用衔接子引物
条形码可以包括一个或多个通用衔接子引物。例如,基因特异性条形码(诸如基因特异性随机条形码)可以包括通用衔接子引物。通用衔接子引物可以指在所有条形码上通用的核苷酸序列。通用衔接子引物可以用于构建基因特异性条形码。通用衔接子引物的长度可以是、或是约1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30个核苷酸、或在这些值的任何两个之间的数字或范围的核苷酸。通用衔接子引物的长度可以是至少、或至多1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、或30个核苷酸。通用衔接子引物的长度可以是5至30个核苷酸。
固体支持物
在一些实施例中,本文披露的条形码(诸如随机条形码)可以与固体支持物相关联。例如,固体支持物可以是合成颗粒。在一些实施例中,固体支持物上的多个条形码(例如,第一多个条形码)的一些或所有条形码序列(诸如,随机条形码(例如,第一条形码序列)的分子标记)具有至少一个核苷酸的差异。相同固体支持物上的条形码的细胞标记可以是相同的。不同的固体支持物上的条形码的细胞标记可以具有至少一个核苷酸的差异。例如,第一固体支持物上的第一多个条形码的第一细胞标记可以具有相同的序列,且第二固体支持物上的第二多个条形码的第二细胞标记可以具有相同的序列。第一固体支持物上的第一多个条形码的第一细胞标记和第二固体支持物上的第二多个条形码的第二细胞标记可以具有至少一个核苷酸的差异。细胞标记可以是例如约5至20个核苷酸长。条形码序列可以是例如约5至20个核苷酸长。合成颗粒可以是例如珠。
珠可以例如是硅胶珠、可控孔径玻璃珠、磁珠、dynabead、交联葡聚糖/琼脂糖珠、珠状纤维素、聚苯乙烯珠、或其任何组合。珠可以包括诸如聚二甲基硅氧烷(pdms)、聚苯乙烯、玻璃、聚丙烯、琼脂糖、明胶、水凝胶、顺磁物质、陶瓷、塑料、玻璃、甲基苯乙烯、丙烯酸聚合物、钛、乳胶、琼脂糖凝胶、纤维素、尼龙、硅酮、或其任何组合的材料。
在一些实施例中,珠可以是高分子微球(例如可变形的珠或凝胶珠),其用条形码或随机条形码功能化(诸如来自10x基因组公司(10xgenomics)(旧金山,加利福尼亚)的凝胶珠)。在一些实现方式中,凝胶珠可以包括基于聚合物的凝胶。例如,可以通过将一种或多种聚合物前体包封进液滴来产生凝胶珠。在将聚合物前体暴露于促进剂(例如,四甲基乙二胺(temed))后,可以产生凝胶珠。
在一些实施例中,颗粒可以是可降解的。例如,高分子微球可以例如在所期望的条件下溶解、熔化或降解。所期望的条件可以包括环境条件。所期望的条件可导致高分子微球以受控方式溶解、熔化或降解。由于化学刺激、物理刺激、生物刺激、热刺激、磁刺激、电刺激、光刺激或其任何组合,凝胶珠可以溶解、融化或降解。
分析物和/或试剂(诸如寡核苷酸条形码)例如可以偶联/固定到凝胶珠的内表面(寡核苷酸条形码和/或用于产生寡核苷酸条形码的材料的扩散的可接近内部)和/或凝胶珠的外表面或本文描述的任何其他微胶囊。偶联/固定可以经由任何形式的化学键(例如,共价键、离子键)或物理现象(例如,范德华力、偶极-偶极相互作用等)。在一些实施例中,试剂与凝胶珠或本文描述的任何其他微胶囊的偶联/固定可以是可逆的,例如经由不稳定部分(例如,经由化学交联剂,包括本文描述的化学交联剂)。在施加刺激后,不稳定部分可以被切割并释放固定化的试剂。在一些实施例中,不稳定部分是二硫键。例如,在经由二硫键将寡核苷酸条形码固定到凝胶珠上的情况下,将二硫键暴露于还原剂可以切割二硫键并从珠释放寡核苷酸条形码。不稳定部分可以作为凝胶珠或微胶囊的一部分、作为将试剂或分析物与凝胶珠或微胶囊连接的化学接头的一部分、和/或作为试剂或分析物的一部分包括在内。在一些实施例中,多个条形码的至少一个条形码可固定在颗粒上、部分固定在颗粒上、包封在颗粒中、部分包封在颗粒中、或其任何组合。
在一些实施例中,凝胶珠可以包括广泛多种不同的聚合物,包括但不限于:聚合物、热敏聚合物、光敏聚合物、磁性聚合物、ph敏感聚合物、盐敏感聚合物、化学敏感聚合物、聚电解质、多糖、肽、蛋白质和/或塑料。聚合物可包括但不限于以下材料:如聚(n-异丙基丙烯酰胺)(pnipaam)、聚(苯乙烯磺酸酯)(pss)、聚(烯丙基胺)(paam)、聚(丙烯酸)(paa)、聚(乙烯亚胺)(pei)、聚(双烯丙基二甲基-氯化铵)(pdadmac)、聚(吡咯)(ppy)、聚(乙烯基吡咯烷酮)(pvpon)、聚(乙烯基吡啶)(pvp)、聚(甲基丙烯酸甲酯)(pmaa)、聚(甲基丙烯酸甲酯)(pmma)、聚苯乙烯(ps)、聚(四氢呋喃)(pthf)、聚(邻苯二甲醛)(pthf)、聚(己基紫精)(phv)、聚(l-赖氨酸)(pll)、聚(l-精氨酸)(parg)、聚(乳酸-聚羟基乙酸)(plga)。
许多化学刺激可用于触发珠的破坏、溶解、或降解。这些化学改变的实例可包括但不限于ph介导的珠壁改变、经由交联键的化学裂解使珠壁分解、珠壁的触发解聚、和珠壁转换反应。批量改变也可用于触发珠的破坏。
通过各种刺激对微胶囊的批量或物理变化在设计胶囊以释放试剂方面也提供了许多优点。在宏观尺度上发生批量或物理变化,其中珠破裂是由刺激引起的机械-物理力的结果。这些过程可包括但不限于压力引起的破裂、珠壁熔化、或珠壁的孔隙率的改变。
生物刺激也可用于触发珠的破坏、溶解、或降解。通常,生物触发剂类似于化学触发剂,但是许多实例使用生物分子、或生命系统中常见的分子,如酶、肽、糖、脂肪酸、核酸等。例如,珠可包括具有肽交联的聚合物,所述肽交联通过特定蛋白酶对切割敏感。更具体地,一个实例可包括含有gflgk肽交联的微胶囊。在添加生物触发物(如蛋白酶组织蛋白酶b)后,壳孔的肽交联被切割且珠的内容物被释放。在其他情况下,蛋白酶可以是热激活的。在另一个实例中,珠包括含有纤维素的壳壁。水解酶壳聚糖的添加用作纤维素键裂解、壳壁解聚、和内部内容物释放的生物触发剂。
还可以在施加热刺激后诱导珠释放其内容物。温度的变化可导致珠的各种变化。热量的变化可能导致珠熔化,使得珠壁崩解。在其他情况下,热量可能增加珠内部组分的内部压力,使得珠破裂或爆炸。在仍其他情况下,热量可以使珠变成收缩的脱水状态。热量还可以作用于珠壁内的热敏聚合物,从而引起珠的破坏。
将磁性纳米颗粒包括在微胶囊的珠壁中可以允许珠的触发破裂以及将珠引导成阵列。本披露的装置可以包括用于任一目的的磁珠。在一个实例中,将fe3o4纳米颗粒并入含聚电解质的珠中在振荡磁场刺激的存在下触发破裂。
由于电刺激的结果,珠也可能被破坏、溶解、或降解。与先前部分中描述的磁性颗粒相似,电敏珠可以允许珠的触发破裂以及其他功能,如电场中的对准、电导率或氧化还原反应。在一个实例中,含有电敏材料的珠在电场中排列,从而可以控制内部试剂的释放。在其他实例中,电场可以在珠壁本身内引起氧化还原反应,这可以增加孔隙率。
也可用光刺激来破坏珠。许多光触发是可能的,并可以包括使用各种分子(如能够吸收特定波长范围的光子的纳米颗粒和发色团)的系统。例如,金属氧化物涂层可用作胶囊触发剂。涂覆有sio2的聚电解质胶囊的uv照射可导致珠壁的崩解。在又另一个实例中,可以将可光切换材料(如偶氮苯基团)并入珠壁中。在施加uv或可见光后,如这些的化学物质在吸收光子后经历可逆的顺式-反式异构化。在此方面,光子切换的并入导致珠壁在施加光触发剂后可崩解或变得更多孔。
例如,在图2中展示的条形码化(例如,随机条形码化)的非限制性实例中,在框208处将细胞(诸如单细胞)引入微孔阵列的多个微孔之后,在框212处可以将珠引入微孔阵列的多个微孔。每个微孔可包括一个珠。珠可以包括多个条形码。条形码可以包括附接到珠的5’胺区域。条形码可以包括通用标记、条形码序列(例如,分子标记)、靶结合区、或其任何组合。
本文披露的条形码可以与固体支持物(例如,珠)相关联(例如,附接)。与固体支持物关联的条形码每个可包括选自下组的条形码序列,该组由以下组成:具有独特序列的至少100或1000个条形码序列。在一些实施例中,与固体支持物相关联的不同条形码可以包括具有不同序列的条形码。在一些实施例中,与固体支持物相关联的条形码的百分比包括相同的细胞标记。例如,所述百分比可以是、或是约60%、70%、80%、85%、90%、95%、97%、99%、100%、或在这些值的任何两个之间的数字或范围。作为另一个实例,所述百分比可以是至少、或至多60%、70%、80%、85%、90%、95%、97%、99%、或100%。在一些实施例中,与固体支持物相关联的条形码可以具有相同的细胞标记。与不同固体支持物相关联的条形码可以具有选自下组的不同的细胞标记,该组由以下组成:具有独特序列的至少100或1000个细胞标记。
本文披露的条形码可以与固体支持物(例如,珠)关联(例如,附接)。在一些实施例中,可以用包括与多个条形码相关联的多个合成的颗粒的固体支持物对样品中的多个靶进行条形码化。在一些实施例中,固体支持物可包括与多个条形码相关联的多个合成的颗粒。不同固体支持物上的多个条形码的空间标记可以具有至少一个核苷酸的差异。固体支持物例如可以包括处于二维或三维的多个条形码。合成的颗粒可以是珠。珠可以是硅胶珠、可控孔径玻璃珠、磁珠、dynabead、交联葡聚糖/琼脂糖珠、珠状纤维素、聚苯乙烯珠、或其任何组合。固体支持物可包括聚合物、基质、水凝胶、针阵列装置、抗体、或其任何组合。在一些实施例中,固体支持物可以自由浮动。在一些实施例中,固体支持物可嵌入半固体或固体阵列中。条形码可以不与固体支持物相关联。条形码可以是单独的核苷酸。条形码可与基底相关联。
如本文使用的,术语“拴系”、“附接”和“固定”可以互换使用,并且可以指用于将条形码附接到固体支持物上的共价或非共价方式。可以将多种不同的固体支持物中的任何一种用作固体支持物,以用于附接预先合成的条形码或用于条形码的原位固相合成。
在一些实施例中,固体支持物是珠。珠可以包括一种或多种类型的实心的、多孔的或空心的球体、球、承座、圆柱体或其他相似配置,其上可以固定核酸(例如,共价地或非共价地)。珠可以例如由塑料、陶瓷、金属、聚合物材料、或其任何组合构成。珠可以是、或包括球形的(例如,微球)或具有非球形或不规则形状的离散颗粒,所述形状是诸如立方形、长方形、锥形、圆柱形、圆锥形、椭圆形或圆盘形等。在一些实施例中,珠的形状可以是非球形的。
珠可以包括多种材料,包括但不限于顺磁性材料(例如,镁、钼、锂和钽)、超顺磁性材料(例如,铁氧体(fe3o4;磁铁矿)纳米颗粒)、铁磁材料(例如,铁、镍、钴,其一些合金,以及一些稀土金属化合物)、陶瓷、塑料、玻璃、聚苯乙烯、二氧化硅、甲基苯乙烯、丙烯酸聚合物、钛、胶乳、交联琼脂糖、琼脂糖、水凝胶、聚合物、纤维素、尼龙、或其任何组合。
在一些实施例中,珠(例如,标记所附接的珠)是水凝胶珠。在一些实施例中,水凝胶珠是可溶解的。在一些实施例中,珠包括水凝胶。
本文披露的一些实施例包括一个或多个颗粒(例如,珠)。颗粒的每个可包括多个寡核苷酸(例如,条形码)。多个寡核苷酸中的每一个可以包括条形码序列(例如,分子标记序列)、细胞标记、和靶结合区(例如,寡聚(dt)序列、基因特异性序列、随机多聚体、或其组合)。所述多个寡核苷酸的每个的细胞标记序列可以是相同的。不同颗粒上的寡核苷酸的细胞标记序列可以是不同的,使得可以鉴定不同颗粒上的寡核苷酸。在不同实现方式中,不同细胞标记序列的数量可以是不同的。在一些实施例中,细胞标记序列的数量可以是、可以是约、可以是至少、或可以是至多10、100、200、300、400、500、600、700、800、900、1000、2000、3000、4000、5000、6000、7000、8000、9000、10000、20000、30000、40000、50000、60000、70000、80000、90000、100000、106、107、108、109、在这些值的任何两个之间的数字或范围、或更多。在一些实施例中,多个颗粒中不超过或不超过约1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900、1000、或更多个包括具有相同细胞序列的寡核苷酸。
在每个颗粒上的多个寡核苷酸可以包括不同的条形码序列(例如,分子标记)。在一些实施例中,条形码序列的数量可以是、可以是约、可以是至少、或可以是至多10、100、200、300、400、500、600、700、800、900、1000、2000、3000、4000、5000、6000、7000、8000、9000、10000、20000、30000、40000、50000、60000、70000、80000、90000、100000、106、107、108、109、在这些值的任何两个之间的数字或范围。作为另一个实例,在单个颗粒中,多个寡核苷酸中的至少100、500、1000、5000、10000、15000、20000、50000个、这些值的任何两个之间的数字或范围、或更多个包括不同的条形码序列。一些实施例提供了包括条形码的多个颗粒。在一些实施例中,待标记的靶和不同条形码序列的出现(或拷贝或数量)的比率可以是至少1:1、1:2、1:3、1:4、1:5、1:6、1:7、1:8、1:9、1:10、1:11、1:12、1:13、1:14、1:15、1:16、1:17、1:18、1:19、1:20、1:30、1:40、1:50、1:60、1:70、1:80、1:90、或更高。在一些实施例中,多个寡核苷酸的每个进一步包括样品标记、通用标记、或两者。颗粒例如可以是纳米颗粒或微颗粒。
条形码化的方法
本披露提供了用于估计身体样品(例如,组织、器官、肿瘤、细胞)中的不同位置处的不同靶的数量的方法。所述方法可以包括将条形码(例如,随机条形码)靠近样品放置,裂解样品,将不同靶与条形码相关联,对所述靶进行扩增和/或对靶进行数字计数。所述方法可以进一步包括对获得自条形码上的空间标记的信息进行分析和/或可视化。在一些实施例中,所述方法包括使样品中的多个靶可视化。将多个靶映射到样品的映射图上可以包括产生样品的二维映射图或三维映射图。可以在对样品中的多个靶进行条形码化(例如,随机条形码化)之前或之后产生二维映射图和三维映射图。将样品中的多个靶可视化包括将多个靶映射到样品的映射图上。将多个靶映射到样品的映射图上可以包括产生样品的二维映射图或三维映射图。可以在对样品中的多个靶进行条形码化之前或之后产生二维映射图和三维映射图。在一些实施例中,可以在裂解样品之前或之后产生二维映射图和三维映射图。在产生二维映射图或三维映射图之前或之后裂解样品可以包括加热样品、使样品与洗涤剂接触、改变样品的ph、或其任何组合。
在一些实施例中,对多个靶进行条形码化包括将多个条形码与多个靶杂交以创建经条形码化的靶(例如,经随机条形码化的靶)。对多个靶进行条形码化可以包括产生经条形码化的靶的索引文库。产生经条形码化的靶的索引文库可以用包括多个条形码(例如,随机条形码)的固体支持物进行。
使样品和条形码接触
本披露提供了用于使样品(例如,细胞)与本披露的基底接触的方法。可以使包括例如细胞、器官或组织薄片的样品与条形码(例如,随机条形码)接触。例如,通过重力流可以使所述细胞接触,其中可以使所述细胞沉淀并且产生单层细胞。所述样品可以是组织薄切片。可以将薄切片置于基底上。所述样品可以是一维的(例如,形成平面)。可以将所述样品(例如,细胞)涂布于基底上,例如,通过在基底上生长/培养所述细胞。
当条形码靠近靶时,靶可以与条形码进行杂交。条形码可以按不可耗尽的比率接触,使得每个不同的靶可以与本披露的不同条形码相关联。为了确保靶与条形码之间的有效关联,可以将靶与条形码交联。
细胞裂解
在细胞和条形码的分布之后,可以裂解细胞以释放靶分子。细胞裂解可以通过多种手段中的任何一种来完成,例如通过化学或生化手段,通过渗透冲击,或通过热裂解、机械裂解或光学裂解。可以通过添加包括洗涤剂(例如,sds、十二烷基硫酸锂、tritonx-100、tween-20或np-40)的细胞裂解缓冲液、有机溶剂(例如甲醇或丙酮)或消化酶(例如蛋白酶k、胃蛋白酶或胰蛋白酶)或其任何组合来裂解细胞。为了增加靶和条形码的关联,可通过例如降低裂解物的温度和/或增加裂解物的粘度来改变靶分子的扩散速率。
在一些实施例中,裂解可以通过机械裂解、热裂解、光学裂解、和/或化学裂解来进行。裂解的细胞可以包括至少约100000、200000、300000、400000、500000、600000、或700000个靶核酸分子,或更多。裂解的细胞可以包括至多约100000、200000、300000、400000、500000、600000、或700000个靶核酸分子,或更多。
将条形码附接到靶核酸分子
在细胞裂解和核酸分子从释放之后,核酸分子可以随机地与共定位的固体支持物的条形码相关联。关联可以包括将条形码的靶识别区与靶核酸分子的互补部分的杂交(例如,条形码的寡聚(dt)可与靶的聚(a)尾相互作用)。可以选择用于杂交的测定条件(例如,缓冲液ph、离子强度、温度等)以促进形成特定的稳定的杂交体。在一些实施例中,可以将从裂解的细胞释放出的核酸分子与基底上的多个探针(例如,与基底上的探针杂交)相关联。当所述探针包括寡聚(dt)时,可以将mrna分子与探针杂交,并且进行逆转录。可以将寡核苷酸的寡聚(dt)部分充当用于cdna分子的第一链合成的引物。例如,图2中(在框216上)说明的条形码化的非限制性实例中,mrna分子可以与珠上的条形码杂交。例如,单链的核苷酸片段可以与条形码的靶结合区杂交。
附接可以进一步包括将条形码的靶识别区与靶核酸分子的一部分连接。例如,靶结合区可以包括可能够与限制性位点突出端(例如,ecori粘性末端突出端)进行特异性杂交的核酸序列。测定程序可以进一步包括用限制性酶(例如,ecori)处理靶核酸以产生限制性位点突出端。然后条形码可以连接到包括与限制性位点突出端互补的序列的任何核酸分子。连接酶(例如,t4dna连接酶)可用于连接两个片段。
例如,在图2(在框220处)中说明的条形码化的非限制性实例中,随后可以将来自多个细胞(或多个样品)的经标记的靶(例如,靶-条形码分子)例如池化至管中。经标记的靶可以通过例如回收条形码和/或附接靶-条形码分子的珠来池化。
可以通过使用磁珠和外部施加的磁场来实现附接的靶-条形码分子的基于固体支持物的集合的检索。一旦所述靶-条形码分子已经池化,所有进一步的处理可以在单个反应容器中进行。进一步的处理可以包括例如逆转录反应、扩增反应、切割反应、解离反应和/或核酸延伸反应。进一步的处理反应可以在微孔内进行,即,不首先池化来自多个细胞的经标记的靶核酸分子。
逆转录
本披露提供了使用逆转录来产生靶-条形码缀合物的方法(在图2的框224中)。靶-条形码缀合物可以包括条形码以及靶核酸(即,条形码化的cdna分子,诸如随机条形码化的cdna分子)的全部或部分的互补性序列。关联的rna分子的逆转录可以通过添加逆转录引物连同逆转录酶一起而发生。逆转录引物可以是寡聚dt引物、随机六核苷酸引物或靶特异性寡核苷酸引物。寡聚(dt)引物的长度可以是、或可以是约12至18个核苷酸,并与哺乳动物mrna的3’端的内源性聚(a)尾结合。随机六核苷酸引物可在多个互补位点处结合至mrna。靶特异性寡核苷酸引物通常选择性地引发目的mrna。
在一些实施例中,标记的rna分子的逆转录可通过添加逆转录引物而进行。在一些实施例中,所述逆转录引物是寡聚(dt)引物、随机六核苷酸引物或靶特异性寡核苷酸引物。通常,寡聚(dt)引物的长度为12至18个核苷酸,并结合至在哺乳动物mrna的3’端的内源性聚(a)尾。随机六核苷酸引物可在多个互补位点处结合至mrna。靶特异性寡核苷酸引物通常选择性地引发目的mrna。
逆转录可以重复地发生以产生多个经标记的cdna分子。本文披露的方法可以包括进行至少约1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20次逆转录反应。所述方法可以包括进行至少约25、30、35、40、45、50、55、60、65、70、75、80、85、90、95或100次逆转录反应。
扩增
可以进行一个或多个核酸扩增反应(例如,在图2的框228中)以产生经标记的靶核酸分子的多个拷贝。扩增能以多路方式进行,其中多个靶核酸序列同时进行扩增。扩增反应可用于向核酸分子添加测序衔接子。扩增反应可以包括扩增样品标记(如果存在)的至少一部分。扩增反应可以包括扩增细胞标记和/或条形码序列(例如,分子标记)的至少一部分。扩增反应可以包括扩增样品标签、细胞标记、空间标记、条形码序列(例如,分子标记)、靶核酸或其组合的至少一部分。扩增反应可包括扩增多个核酸的0.5%、1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、97%、100%、或在这些值的任何两个之间的数字或范围。所述方法可以进一步包括进行一个或多个cdna合成反应以产生包括样品标记、细胞标记、空间标记和/或条形码序列(例如,分子标记)的靶-条形码分子的一个或多个cdna拷贝。
在一些实施例中,可以使用聚合酶链式反应(pcr)进行扩增。如本文使用的,pcr可以指用于通过dna的互补链的同时引物延伸使特异性dna序列体外扩增的反应。如本文使用的,pcr可以包括所述反应的派生形式,包括但不限于rt-pcr、实时pcr、巢式pcr、定量pcr、多重pcr、数字pcr、和组装pcr。
经标记的核酸的扩增可以包括基于非pcr的方法。非基于pcr的方法的实例包括但不限于多重置换扩增(mda)、转录介导的扩增(tma)、基于核酸序列的扩增(nasba)、链置换扩增(sda)、实时sda、滚环扩增或环到环扩增(circle-to-circleamplification)。其他非基于pcr的扩增方法包括dna依赖性rna聚合酶驱动的rna转录扩增或rna指导的dna合成和转录的多个循环以扩增dna或rna靶、连接酶链式反应(lcr)、和qβ复制酶(qβ)方法、回文探针的使用、链置换扩增、使用限制性内切核酸酶的寡核苷酸驱动的扩增、使引物与核酸序列杂交并且将所得双链体在延伸反应和扩增之前切割的扩增方法、使用缺乏5’外切核酸酶活性的核酸聚合酶的链置换扩增、滚环扩增和分支延伸扩增(ram)。在一些实施例中,扩增不产生环化转录物。
在一些实施例中,本文披露的方法进一步包括对经标记的核酸(例如,经标记的rna、经标记的dna、经标记的cdna)进行聚合酶链式反应,以产生经标记的扩增子(例如,经随机标记的扩增子)。所述经标记的扩增子可以是双链分子。双链分子可包括双链rna分子、双链dna分子或者与dna分子杂交的rna分子。双链分子的一条或两条链可以包括样品标记、空间标记、细胞标记、和/或条形码序列(例如,分子标记)。所述经标记的扩增子可以是单链分子。单链分子可包括dna、rna或其组合。本披露的核酸可以包括合成的或改变的核酸。
扩增可以包括使用一个或多个非天然核苷酸。非天然核苷酸可以包括光不稳定或可触发的核苷酸。非天然核苷酸的实例可以包括但不限于肽核酸(pna)、吗啉代和锁核酸(lna)、以及二醇核酸(gna)与苏糖核酸(tna)。可以将非天然核苷酸添加至扩增反应的一个或多个循环中。添加非天然核苷酸也可以用于鉴别扩增反应中特定循环或时间点的产物。
一个或多个引物可以包括通用引物。通用引物可以退火至通用引物结合位点。一个或多个定制引物可以退火至第一样品标记、第二样品标记、空间标记、细胞标记、条形码序列(例如,分子标记)、靶、或其任何组合。一个或多个引物可以包括通用引物和定制引物。定制引物可以设计用于扩增一个或多个靶。靶可以包括一个或多个样品中总核酸的子集。靶可以包括一个或多个样品中总的经标记的靶的子集。一个或多个引物可以包括至少96个或更多个定制引物。一个或多个引物可以包括至少960个或更多个定制引物。一个或多个引物可以包括至少9600个或更多个定制引物。一个或多个定制引物可以退火至两个或更多个不同的经标记的核酸。两个或更多个不同的经标记的核酸可以对应于一个或多个基因。
可以在本披露的方法中使用任何扩增方案。例如,在一个方案中,第一轮pcr可以使用基因特异性引物和针对通用亿明达(illumina)测序引物1序列的引物来扩增附接到珠上的分子。第二轮pcr可以使用侧翼于亿明达测序引物2序列的巢式基因特异性引物和针对通用亿明达测序引物1序列的引物扩增第一pcr产物。第三轮pcr添加p5和p7以及样品索引,以便使pcr产物进入亿明达测序文库。使用150bpx2测序的测序可以揭示读数1上的细胞标记和条形码序列(例如,分子标记)、读数2上的基因、以及索引1读数上的样品索引。
在一些实施例中,使用化学切割可以将核酸从基底中去除。例如,可以将存在于核酸中的化学基团或经修饰的碱基用于促进将其从固体支持物中去除。例如,酶可以用于从基底中去除核酸。例如,通过限制性内切核酸酶消化,可以将核酸从基底中去除。例如,使用尿嘧啶-d-糖基化酶(udg)处理含有dutp或ddutp的核酸可以从基底中去除核酸。例如,可以使用用于核苷酸切除(例如,碱基切除修复酶(例如,脱嘌呤/脱嘧啶(ap)核酸内切酶))的酶将核酸从基底中去除。在一些实施例中,可以使用可光解(photocleavable)基团以及光将核酸从基底中去除。在一些实施例中,可以使用可切割接头从基底中去除核酸。例如,可切割接头可以包括以下中的至少一种:生物素/亲和素、生物素/链霉抗生物素蛋白、生物素/中性链亲和素、ig蛋白a、光不稳定性接头、酸或碱不稳定性接头基团、或适配体。
当探针是基因特异性时,可以将所述分子与探针杂交,并且进行逆转录和/或扩增。在一些实施例中,在核酸已经合成(例如,逆转录)之后,可以将其扩增。扩增能以多重方式进行,其中多个靶核酸序列同时进行扩增。扩增可以将测序衔接子添加至核酸。
在一些实施例中,可以例如用桥接扩增在基板上进行扩增。cdna可以加同聚物尾,以便产生相容末端用于使用基底上的寡聚(dt)探针进行桥接扩增。在桥接扩增中,与模板核酸的3’端互补的引物可以是共价附接到固体颗粒的每对引物的第一引物。当含有模板核酸的样品与颗粒接触并进行单个热循环时,可以将模板分子退火至第一引物,并且第一引物通过添加核苷酸而向前延伸以形成双链体分子,所述双链体分子由模板分子和与模板互补的新形成的dna链构成。在下一循环的加热步骤中,双链体分子可以变性,从颗粒释放模板分子,并通过第一引物将互补性dna链附接至颗粒。在随后的退火和延伸步骤的退火阶段中,互补链可以与第二引物杂交,所述第二引物在从第一引物去除的位置处与互补链的片段互补。所述杂交可导致互补链在通过共价键固定到第一引物的第一和第二引物之间形成桥接,并通过杂交形成第二引物。在延伸阶段,通过在相同的反应混合物中添加核苷酸,第二引物可以按相反方向延伸,从而将桥转化为双链桥。然后开始下一个循环,并且所述双链桥可以变性以产生两个单链核酸分子,每个单链核酸分子的一端分别经第一和第二引物附接至颗粒表面,其中每个单链核酸分子的另一端是未附接的。在所述第二个循环的退火和延伸步骤中,每条链可以与先前未使用的另外的互补引物杂交在相同的颗粒上,以形成新的单链桥。将现在杂交的两个先前未使用的引物延伸从而将两个新桥转换成双链桥。
对经标记的核酸的扩增可以包括基于pcr的方法或非基于pcr的方法。对经标记的核酸的扩增可以包括对经标记的核酸的索引式扩增。对经标记的核酸的扩增可以包括对经标记的核酸的线性扩增。扩增可以通过聚合酶链式反应(pcr)来进行。pcr可指用于通过dna的互补链的同时引物延伸使特异性dna序列体外扩增的反应。pcr可涵盖所述反应的派生形式,包括但不限于,rt-pcr、实时pcr、巢式pcr、定量pcr、多重pcr、数字pcr、抑制pcr、半抑制pcr以及装配pcr。
在一些实施例中,所述经标记的核酸的扩增包括非基于pcr的方法。非基于pcr的方法的实例包括但不限于多重置换扩增(mda)、转录介导的扩增(tma)、基于核酸序列的扩增(nasba)、链置换扩增(sda)、实时sda、滚环扩增或环到环扩增(circle-to-circleamplification)。其他非基于pcr的扩增方法包括dna依赖性rna聚合酶驱动的rna转录扩增或rna指导的dna合成和转录的多个循环以扩增dna或rna靶、连接酶链式反应(lcr)、qβ复制酶(qβ)、回文探针的使用、链置换扩增、使用限制性内切核酸酶的寡核苷酸驱动的扩增、使引物与核酸序列杂交并且将所得双链体在延伸反应和扩增之前切割的扩增方法、使用缺乏5’外切核酸酶活性的核酸聚合酶的链置换扩增、滚环扩增和分支延伸扩增(ram)。
在一些实施例中,本文披露的所述方法进一步包括对扩增的扩增子(例如,靶)进行巢式聚合酶链式反应。扩增子可以是双链分子。双链分子可包括双链rna分子、双链dna分子或者与dna分子杂交的rna分子。双链分子的一条或两条链可包括样品标签或分子鉴定物标记。可替代地,所述扩增子可以是单链分子。单链分子可包括dna、rna或其组合。本发明的核酸可以包括合成的或改变的核酸。
在一些实施例中,所述方法包括反复扩增经标记的核酸以产生多个扩增子。本文披露的方法可包括进行至少约1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20次扩增反应。可替代地,所述方法包括进行至少约25、30、35、40、45、50、55、60、65、70、75、80、85、90、95或100次扩增反应。
扩增可进一步包括将一个或多个对照核酸添加至一个或多个包括多个核酸的样品中。扩增可进一步包括将一个或多个对照核酸添加至多个核酸中。对照核酸可以包括对照标记。
扩增可以包括使用一个或多个非天然核苷酸。非天然核苷酸可以包括光不稳定和/或可触发的核苷酸。非天然核苷酸的实例包括但不限于肽核酸(pna)、吗啉代和锁核酸(lna)以及二醇核酸(gna)与苏糖核酸(tna)。可以将非天然核苷酸添加至扩增反应的一个或多个循环中。添加非天然核苷酸也可以用于鉴别扩增反应中特定循环或时间点的产物。
进行一个或多个扩增反应可以包括使用一个或多个引物。一个或多个引物可以包括一个或多个寡核苷酸。一个或多个寡核苷酸可以包括至少约7至9个核苷酸。一个或多个寡核苷酸可以包括少于12至15个核苷酸。一个或多个引物可以退火至多个经标记的核酸的至少一部分。一个或多个引物可以退火至多个经标记的核酸的3’端和/或5’端。一个或多个引物可以退火至多个经标记的核酸的内部区。内部区可以是从所述多个经标记的核酸的3’端的至少约50、100、150、200、220、230、240、250、260、270、280、290、300、310、320、330、340、350、360、370、380、390、400、410、420、430、440、450、460、470、480、490、500、510、520、530、540、550、560、570、580、590、600、650、700、750、800、850、900或1000个核苷酸。一个或多个引物可以包括一组固定的引物。一个或多个引物可以包括至少一个或多个定制引物。一个或多个引物可以包括至少一个或多个对照引物。一个或多个引物可以包括至少一个或多个管家基因引物。一个或多个引物可以包括通用引物。通用引物可以退火至通用引物结合位点。一个或多个定制引物可以退火至第一样品标签、第二样品标签、分子鉴定物标记、核酸或它们的产物。一个或多个引物可以包括通用引物和定制引物。定制引物可以被设计成扩增一个或多个靶核酸。靶核酸可以包括一个或多个样品中总核酸的子集。在一些实施例中,所述引物是附接至本披露的阵列的探针。
在一些实施例中,使样品中的多个靶条形码化(例如,随机条形码化)进一步包括生成经条形码化的靶(例如,经随机条形码化的靶)的索引文库或所述靶的经条形码化的片段的索引文库。不同的条形码的条形码序列(例如,不同的随机条形码的分子标记)可以彼此不同。生成经条形码化的靶的索引文库包括从样品中的多个靶生成多个索引多核苷酸。例如,对于包括第一索引靶和第二索引靶的经条形码化的靶的索引文库,第一索引多核苷酸的标记区与第二索引多核苷酸的标记区可以具有、具有约、具有至少、或具有至多1、2、3、4、5、6、7、8、9、10、20、30、40、50个核苷酸的差异、或在这些值的任何两个之间的数字或范围的核苷酸差异。在一些实施例中,产生经条形码化的靶的索引文库包括使多个靶(例如mrna分子)与包括聚(t)区和标记区的多个寡核苷酸接触;以及使用逆转录酶进行第一链合成以产生单链标记的cdna分子(每个包括cdna区和标记区),其中多个靶包括不同序列的至少两个mrna分子,且多个寡核苷酸包括不同序列的至少两个寡核苷酸。产生经条形码化的靶的索引文库可进一步包括扩增单链标记的cdna分子以产生双链标记的cdna分子;以及在双链标记的cdna分子上进行巢式pcr以产生标记的扩增子。在一些实施例中,所述方法可包括产生衔接子标记的扩增子。
条形码化(例如,随机条形码化)可以包括使用核酸条形码或标签以标记单个核酸(例如,dna或rna)分子。在一些实施例中,其涉及将dna条形码或标签添加至cdna分子,因为它们是从mrna产生的。可以进行巢式pcr以最小化pcr扩增偏差。可以使用例如下一代测序(ngs)添加衔接子用于测序。例如在图2的框232处,可以使用测序结果以确定靶的一个或多个拷贝的细胞标记、分子标记、和核苷酸片段的序列。
图3是示出了生成经条形码化的靶(例如,经随机条形码化的靶)的索引文库,诸如经条形码化的mrna或其片段的索引文库的非限制性示例性过程的示意图。如步骤1显示,逆转录过程可以编码具有独特的分子标记、细胞标记和通用pcr位点的每个mrna分子。具体地,通过将一组条形码(例如,随机条形码)310杂交(例如,随机杂交)到rna分子302的聚(a)尾区308,可以将rna分子302逆转录以产生经标记的cdna分子304(包括cdna区306)。条形码310中的每一个可以包括靶结合区,例如聚(dt)区312、标记区314(例如,条形码序列或分子)和通用pcr区316。
在一些实施例中,细胞标记可以包括3至20个核苷酸。在一些实施例中,分子标记可以包括3至20个核苷酸。在一些实施例中,多个随机条形码的每个进一步包括通用标记和细胞标记的一个或多个,其中通用标记对于固体支持物上的多个随机条形码是相同的且细胞标记对于固体支持物上的多个随机条形码是相同的。在一些实施例中,通用标记可以包括3至20个核苷酸。在一些实施例中,细胞标记包括3至20个核苷酸。
在一些实施例中,标记区314可包括条形码序列或分子标记318和细胞标记320。在一些实施例中,标记区314可包括通用标记、维度标记、和细胞标记的一个或多个。条形码序列或分子标记318的长度可以是、可以是约、可以是至少、或可以是至多1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100个核苷酸、或在这些值的任何之间的数字或范围的核苷酸。细胞标记320的长度可以是、可以是约、可以是至少、或可以是至多1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100个核苷酸、或在这些值的任何之间的数字或范围的核苷酸。通用标记的长度可以是、可以是约、可以是至少、或可以是至多1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100个核苷酸、或在这些值的任何之间的数字或范围的核苷酸。对于固体支持物上的多个随机条形码,通用标记可以是相同的,且对于固体支持物上的多个随机条形码,细胞标记是相同的。维度标记的长度可以是、可以是约、可以是至少、或可以是至多1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100个核苷酸、或在这些值的任何之间的数字或范围的核苷酸。
在一些实施例中,标记区314可包括、包括约、包括至少、或包括至多1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900、1000个不同标记、或在这些值的任何之间的数字或范围的不同标记,如条形码序列或分子标记318和细胞标记320。每个标记的长度可以是、可以是约、可以是至少、或可以是至多1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100个核苷酸、或在这些值的任何之间的数字或范围的核苷酸。一组条形码或随机条形码310可以含有、含有约、含有至少、或可以是至多10、20、40、50、70、80、90、102、103、104、105、106、107、108、109、1010、1011、1012、1013、1014、1015、1020个条形码或随机条形码310、或在这些值的任何之间的数字或范围的条形码或随机条形码310。并且条形码或随机条形码310的组可以例如,各自含有独特标记区314。经标记的cdna分子304可以进行纯化以去除过量条形码或随机条形码310。纯化可以包括ampure珠纯化。
如步骤2所示,来自逆转录过程的产物在步骤1中可以池化至1管中,且用第1pcr引物池和第1通用pcr引物进行pcr扩增。因为独特标记区314,池化是可能的。特别地,可以将标记的cdna分子304扩增以产生巢式pcr标记的扩增子322。扩增可以包括多重pcr扩增。扩增可以包括在单一反应体积中用96种多重引物进行的多重pcr扩增。在一些实施例中,在单一反应体积中,多重pcr扩增可以利用、利用约、利用至少、或利用至多10、20、40、50、70、80、90、102、103、104、105、106、107、108、109、1010、1011、1012、1013、1014、1015、1020个多重引物、或在这些值的任何之间的数字或范围的多重引物。扩增可以包括使用包括靶向特异性基因的定制引物326a-c和通用引物328的第1pcr引物池324。定制引物326可以与经标记的cdna分子304的cdna部分306’内的区域杂交。通用引物328可以与经标记的cdna分子304的通用pcr区域316杂交。
如图3的步骤3中显示,来自步骤2中的pcr扩增的产物可以用巢式pcr引物池和第2通用pcr引物进行扩增。巢式pcr可以最小化pcr扩增偏差。特别地,巢式pcr标记的扩增子322可通过巢式pcr进行进一步扩增。巢式pcr可以包括在单个反应体积中用巢式pcr引物332a-c的巢式pcr引物池330和第2通用pcr引物328’的多重pcr。巢式pcr引物池328可含有、含有约、含有至少、或含有至多1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900、1000个不同巢式pcr引物330、或在这些值的任何之间的数字或范围的不同巢式pcr引物330。巢式pcr引物332可含有衔接子334,并与经标记的扩增子322的cdna部分306”内的区域杂交。通用引物328’可含有衔接子336,并与经标记的扩增子322的通用pcr区域316杂交。因此,步骤3产生衔接子标记的扩增子338。在一些实施例中,巢式pcr引物332和第2通用pcr引物328’可以不含有衔接子334和336。相反,衔接子334和336可以连接到巢式pcr的产物以产生衔接子标记的扩增子338。
如步骤4中显示,可以使用文库扩增引物将来自步骤3的pcr产物进行pcr扩增用于测序。特别地,可以将衔接子334和336用于对衔接子标记的扩增子338执行一个或多个另外的测定。衔接子334和336可以与引物340和342杂交。一个或多个引物340和342可以是pcr扩增引物。一个或多个引物340和342可以是测序引物。一个或多个衔接子334和336可以用于衔接子标记的扩增子338的进一步扩增。一个或多个衔接子334和336可以用于对衔接子标记的扩增子338进行测序。引物342可含有板索引344,使得使用同一组条形码或随机条形码310产生的扩增子可以使用下一代测序(ngs)在一轮测序反应中测序。
测序数据错误
本文披露的方法可以用于鉴定和/或校正测序数据错误,例如在用于计数一个或多个靶核酸的方法中发生的错误。在一些实施例中,错误可以包括、或是一个或多个核苷酸的缺失、一个或多个核苷酸的取代、和一个或多个核苷酸的添加、或其任何组合。错误可以存在于分子标记(ml)、样品标记(sl)、或条形码(例如,随机条形码)上的其他标记上。在一些实施例中,测序数据错误可以包括、或是pcr引入的错误、测序引入的错误、逆转录(rt)引物污染错误、或其任何组合。pcr引入的错误可包括、或是pcr扩增错误、pcr扩增偏差、pcr扩增不足、或其任何组合的结果。测序引入的错误可包括、或是不精确的碱基调用、测序不足、或其任何组合的结果。rt引物污染错误可以是由进入pcr的逆转录引物引起的错误。
如本文所用的,术语“覆盖”或“测序深度”可以指在测序数据中具有特定ml和特定sl的条形码化的靶的读数。例如,可以对条形码化的靶多次测序。因此,可以多次观察具有特定ml和sl的条形码化的靶。作为另一个实例,细胞可以含有靶的多个拷贝(例如,基因的mrna分子的多个拷贝)。靶的多个拷贝可以被条形码化。在pcr扩增(例如,图2中的框228)后,可以有具有特定ml和sl的经条形码化的靶的多个拷贝。在测序期间,可以对具有特定ml和sl的条形码化的靶的多个拷贝的一些或所有进行测序。在测序数据中观察到的具有相同ml和sl的条形码化的靶的读数的数量可以称为“覆盖”或“测序深度”。
在一些实施例中,可以鉴定和/或校正测序数据错误。例如,来自细胞的靶的拷贝可以用不同ml和相同sl的条形码化。具有ml的条形码化的靶可以在测序数据中具有多个读数。具有不同ml的条形码化的靶可以仅具有少量读数(例如,一次读数)。与后者条形码化的靶相比,前者条形码化的靶更可能具有真ml(或真实ml或信号ml)。后者条形码化的靶可包括错误ml(或假ml或噪音ml)。这可能是因为可以预期两个ml具有相似的覆盖或测序深度。仅具有少量读数的后者条形码化的靶可以是在测序或pcr期间产生的伪影或错误。
作为另一个实例,进入pcr的条形码(例如,随机条形码)可以导致rt引物污染错误。在一些实施例中,在将mrna分子逆转录为cdna分子之后(例如,在图2的框224处),可以通过例如ampure珠纯化去除未并入cdna分子的条形码。去除方法(例如,ampure珠纯化)可能不能完全去除未通过逆转录延伸以并入经条形码化的cdna分子(例如,经随机条形码化的cdna分子)中的条形码。例如,15%、10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.5%、0.1%、或在这些两个值的任何之间的范围的未通过逆转录延伸以并入经条形码化的cdna分子中的条形码不能通过ampure珠纯化去除。这些未去除的条形码可能导致cdna分子扩增期间(例如,在图2的框228处)的测序数据错误。样品之间的条形码可以是高度相似的。例如,条形码的样品标记对于样品可以是相同的。因此,可以发生pcr交叉,因为在pcr期间这些未去除的条形码可以与来自相同样品的其他核酸分子(例如,经条形码化的mrna分子(诸如经随机条形码化的mrna分子)的sl区域)杂交并且可以导致称为sl错误的测序数据错误。
真ml、错误ml、和sl错误可以具有不同的分布。图4是示出了分子标记错误、样品标记错误、和真分子标记信号的非限制性示例性分布的示意图。如图4中说明的,错误ml可更可能具有较低的ml覆盖,因为错误ml可以是pcr或排序错误的结果。例如,错误ml可以是大多数测序错误和一些pcr错误的结果。sl错误可能更有可能具有较低的ml覆盖,因为sl错误可能主要来自进入pcr的条形码(例如,随机条形码)。
基于定向邻接校正pcr和测序错误
本文披露的是用于校正pcr或测序错误的方法。在一些实施例中,所述方法包括:(a)接收经条形码化的靶(例如,经随机条形码化的靶)的测序数据。可以通过以下获得经条形码化的靶:使用多个条形码(例如,随机条形码)使多个靶条形码化(例如,随机条形码化)以创建多个经条形码化的靶(例如,经随机条形码化的靶),其中,所述多个条形码中的每一个包括分子标记。在一些实施例中,所述方法包括:(b)对于所述多个靶中的一个或多个:(i)对具有与测序数据中的靶相关联的不同序列的分子标记的数量进行计数;(ii)使用定向邻接鉴定所述靶的分子标记的簇;(iii)使用(ii)中鉴定的所述靶的分子标记的簇对(b)中接收的测序数据进行折叠;以及(iv)估计靶的数量,其中,在对(ii)中的测序数据进行折叠后,所述经估计的靶的数量与在(i)中计数的具有跟测序数据中的靶相关联的不同序列的分子标记的数量相关。所述多个靶可以包括细胞的整个转录组的靶。在一些实施例中,所述方法进一步包括:(c)使用所述多个条形码使所述多个靶条形码化(例如,随机条形码化)以创建所述多个经条形码化的靶;以及(d)对所述经条形码化的靶进行测序以生成接收的经随机条形码化的靶的测序数据。
图5是示出了基于定向邻接使用分子标记校正pcr和测序错误的非限制性示例性实施例500的流程图。使用基于定向邻接的分子标记校正pcr和测序错误可以被称为递归取代错误校正(rsec)。在接受多个经条形码化的靶(例如,经随机条形码化的靶)的测序数据后,在框504处开始方法500。在一些实施例中,方法500进一步包括使用多个条形码(例如,随机条形码)使多个靶随机条形码化以创建多个经条形码化的靶(例如,经随机条形码化的靶),其中,所述多个条形码中的每一个包括分子标记。在一些实施例中,方法500进一步包括对多个经条形码化的靶进行测序以获得测序数据。
在框508处,对于所述多个靶中的一个或多个:可以对具有与测序数据中的靶相关联的不同序列的分子标记的数量进行计数。在框512处,使用定向邻接可以鉴定靶的分子标记的簇。簇中的靶的分子标记可以在彼此的预定的定向邻接阈值内。定向邻接阈值可以变化。在一些实施例中,预定的定向邻接阈值的汉明距离可以是、是约、至少、或至多一或二。
在一些实施例中,簇内的靶的分子标记可包括一个或多个亲本分子标记以及所述一个或多个亲本分子标记的子分子标记。亲本分子标记的出现可以大于或等于预定的定向邻接出现阈值。在一些实施例中,预定的定向邻接出现阈值可以是、是约、至少、或至多少于一的子分子标记的出现的两倍。在一些实施例中,预定的定向邻接出现阈值可以是、或是约子分子标记的出现的1.5倍、2倍、3倍、4倍、5倍、6倍、7倍、8倍、9倍、10倍、或在这些值的任何两个之间的数字或范围。在一些实施例中,预定的定向邻接出现阈值可以是子分子标记的出现的至少或至多1.5倍、2倍、3倍、4倍、5倍、6倍、7倍、8倍、9倍、或10倍。
在框520中,使用靶的分子标记的簇将测序数据进行折叠。折叠测序数据可包括将子分子标记的出现归因于亲本分子标记。在框532处,可以估计靶的数量以在对测序数据进行折叠之后产生输出。所述方法在500在框536处结束。
在一些实施例中,所述方法进一步包括:确定靶的测序深度。如果所述靶的测序深度高于预定的测序深度阈值,则对靶的数量进行估计包括调整(i)中计数的测序数据。预定的测序深度阈值可以在15与20之间。对(i)中计数的测序数据进行调整包括:限定靶的分子标记的阈值以确定与(b)中获得的测序数据中的靶相关联的真分子标记和假分子标记。限定靶的分子标记的阈值包括对靶的分子标记进行统计分析。进行所述统计分析包括:将靶的分子标记及其出现的分布拟合至两个分布,如两个负二项分布;使用这两个负二项分布来确定真分子标记的数量n;以及从(b)中获得测序数据中去除假分子标记,其中,所述假分子标记包括具有其出现低于第n个最丰富分子标记的出现的分子标记,并且其中,所述真分子标记包括具有其出现大于或等于第n个最丰富分子标记的出现的分子标记。
基于定向邻接和基于分布的错误校正来校正pcr和测序错误
本文披露的是用于校正pcr或测序错误的方法。可以使用所述方法来确定靶的数量。在一些实施例中,所述方法包括:(a)接收经条形码化的靶(例如,经随机条形码化的靶)的测序数据。可以通过以下获得经条形码化的靶:使用多个条形码(例如,随机条形码)使多个靶条形码化(例如,随机条形码化)以创建多个经条形码化的靶(例如,经随机条形码化的靶),其中,所述多个条形码中的每一个包括分子标记。在一些实施例中,所述方法包括(b)对于多个靶的一个或多个:(i)对具有与测序数据中的靶相关联的不同序列的分子标记的数量进行计数;(ii)确定具有测序数据中的与所述靶相关联的不同序列的噪音分子标记的数量;以及(iii)估计靶的数量,其中,所述经估计的靶的数量与在(i)中计数的具有跟测序数据中的靶相关联的不同序列的分子标记的数量相关,所述分子标记的数量根据(ii)中确定的噪音分子标记的数量调整。在一些实施例中,所述方法包括确定测序数据中的靶的测序状态。在一些实施例中,所述方法进一步包括:(c)使用多个条形码使多个靶条形码化(例如,随机条形码化)以创建多个经条形码化的靶;以及(d)对经条形码化的靶进行测序以生成接收的经条形码化的靶的测序数据。
图6是示出了基于递归取代错误校正和基于分布的错误校正来校正pcr和测序错误的非限制性示例性实施例600的流程图。在接受多个经条形码化的靶(例如,经随机条形码化的靶)的测序数据后,在框604处开始方法600。在一些实施例中,方法600进一步包括使用多个条形码(例如,随机条形码)使多个靶条形码化(例如,随机条形码化)以创建多个经条形码化的靶,其中,所述多个条形码中的每一个包括分子标记。在一些实施例中,方法600进一步包括对多个经条形码化的靶进行测序以获得测序数据。
在框608处,对于所述多个靶中的一个或多个:可以对具有与测序数据中的靶相关联的不同序列的分子标记的数量进行计数。在决策框612处,可以确定测序数据是否具有饱和测序状态。例如,如果靶具有大于1000、2000、3000、4000、5000、6000、7000、8000、9000、10000、20000、30000、40000、50000、60000、70000、80000、90000、100000、或这些的任何两个之间的数字或范围的具有不同序列的分子标记的数量,则所述靶被认为具有饱和的测序状态。作为另一个实例,如果靶具有的具有不同序列的分子标记的数量大于具有不同序列的条形码(例如,随机条形码)的分子标记的50%、60%、70%、80%、90%、95%、99%、99.9%、或在这些的任何两个之间的数字或范围,则可以认为所述靶具有饱和测序状态。
在一些实施例中,饱和的测序状态可以通过具有大于预定的饱和阈值的数量的分子标记的靶来确定,所述分子标记具有不同序列。在不同实现方式中,预定的饱和阈值可以是不同的。例如,预定的饱和阈值可以是、或是约1000、2000、3000、4000、5000、6000、6557、7000、8000、9000、10000、20000、30000、40000、50000、60000、65532、70000、80000、90000、100000、或在这些值的任何两个之间的数字或范围。作为另一个实例,预定的饱和阈值可以是至少、或至多1000、2000、3000、4000、5000、6000、6557、7000、8000、9000、10000、20000、30000、40000、50000、60000、65532、70000、80000、90000、或100000。
在一些实施例中,饱和测序状态可以取决于条形码(例如,随机条形码)的具有不同序列的分子标记的数量。例如,如果条形码包括具有不同序列的约6561个分子标记,则预定的饱和阈值可以是约6557。作为另一个实例,如果条形码(例如,随机条形码)包括具有不同序列的约65536个分子标记,则预定的饱和阈值可以是约65532。在一些实施例中,饱和测序状态可以不取决于条形码的具有不同序列的分子标记的数量。
如果测序数据在决策框612不具有饱和测序状态,则方法600前进到框616处,其中可以基于定向邻接调整分子标记计数。在一些实施例中,基于定向邻接调整分子标记计数可以参考图5描述。例如,基于定向邻接调整分子标记计数可以包括使用定向邻接来鉴定靶的分子标记的簇;使用鉴定的靶的分子标记的簇对测序数据进行折叠;以及估计靶的数量,其中在对测序数据进行折叠后,所述经估计的靶的数量与分子标记的数量关联,所述分子标记具有计数的测序数据中的靶相关联的不同的序列。
在框620处,可以确定测序数据中的靶的测序状态。测序数据中的靶的测序状态可包括、或是测序不足。在决策框624处,可以确定测序数据中的靶的测序状态是否是测序不足状态。例如,如果靶的深度(例如平均、最小、或最大深度)小于、或小于约1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100、或在这些的任何两个之间的数字或范围,则可以认为所述靶具有测序不足状态。作为另一个实例,如果靶的深度小于至少、或至多1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、或100,则可以认为所述靶具有测序不足状态。
在一些实施例中,测序不足状态可以通过具有小于预定的测序不足阈值的深度(例如,平均、最小、或最大深度)的靶来确定。在不同实现方式中,测序不足阈值可以是不同的。例如,测序不足阈值可以是、或是约1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100、或或在这些值的任何两个之间的数字或范围。作为另一个实例,测序不足阈值可以是至少、或至多1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、或100。
在一些实施例中,测序不足状态可以取决于条形码(例如,随机条形码)的具有不同序列的分子标记的数量。例如,如果条形码包括、或约1000、2000、3000、4000、5000、6000、6561、7000、8000、9000、10000、20000、30000、40000、50000、60000、65532、70000、80000、90000、100000个具有不同序列的分子标记、或在这些值的任何两个之间的数字或范围的具有不同序列的分子标记,则测序不足阈值可以是10(或另一个阈值数量)。作为另一个实例,如果条形码包括至少、或至多1000、2000、3000、4000、5000、6000、6561、7000、8000、9000、10000、20000、30000、40000、50000、60000、65532、70000、80000、90000、或100000,则测序不足阈值可以是10(或另一个阈值数量)。在一些实施例中,测序不足状态可以不取决于条形码(例如,随机条形码)的具有不同序列的分子标记的数量。
在决策框624处,如果测序数据中的靶的测序状态不是测序不足状态,则方法600可以前进到框628以过滤分子标记计数。过滤分子标记计数可包括,在决策框632处确定小于伪点阈值的具有与测序数据中的靶相关联的不同序列的分子标记的数量。在不同实现方式中,伪点阈值可以是不同的。例如,如果条形码(例如,随机条形码)包括具有不同序列的约6561个分子标记,则伪点阈值可以是、或是约1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100、或在这些值的任何两个之间的数字或范围。作为另一个实例,如果条形码(例如,随机条形码)包括具有不同序列的约6561个分子标记,则伪点测序阈值可以是至少、或至多1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、或100。
在决策框632处,如果具有与测序数据中的靶相关联的不同序列的分子标记的数量小于伪点阈值,则方法600可任选地前进到框636处,其中在确定具有与测序数据中的靶相关联的不同序列的噪音分子标记的数量之前,可以将伪点添加至具有与测序数据中的靶相关联的不同序列的分子标记的数量中。在不同实现方式中,伪点可以具有不同分子标记计数。例如,伪点的分子标记计数可以是、或是约0.0001、0.001、0.01、0.1、1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100、或或在这些值的任何两个之间的数字或范围。作为另一个实例,伪点的分子标记计数可以是至少、或至多0.0001、0.001、0.01、0.1、1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、或100。
在决策框632处,如果具有与测序数据中的靶相关联的不同序列的分子标记的数量不是小于伪点阈值,则可以在框640处去除非独特分子标记。在框644处,可以通过确定具有测序数据中的与所述靶相关联的不同序列的噪音分子标记的数量来去除非独特分子标记。非独特分子标记可包括具有大于预定的再循环分子标记阈值的与测序数据中的靶相关联的不同序列的分子标记。在不同实现方式中,再循环分子标记阈值可以是不同的。例如,如果条形码(例如,随机条形码)包括具有不同序列的约6561个分子标记,则再循环分子标记阈值可以是、或是约100、200、300、400、500、600、650、700、900、1000、2000、或在这些值的任何两个之间的数字或范围。作为另一个实例,如果条形码(例如,随机条形码)包括具有不同序列的约6561个分子标记,则再循环分子标记阈值可以是至少、或至多100、200、300、400、500、600、650、700、900、1000、或2000。
在一些实施例中,去除非独特分子标记包括:针对具有所述测序数据中的与靶相关联的不同序列的分子标记的数量确定非独特分子标记的理论数量。去除非独特分子标记可包括去除分子标记,该分子标记的出现大于具有与该测序数据中的靶相关联的不同序列的分子标记的第n个最丰富分子标记。该数字n可以是非独特分子标记的理论数。
在框644处,可以使用基于分布的错误校正方法来调整分子标记计数。基于分布的错误校正方法可包括确定具有与测序数据中的靶相关联的不同序列的噪音分子标记的数量。确定噪音分子标记的数量可包括:将两个负二项分布拟合至具有与测序数据中的靶相关联的不同序列的分子标记的数量。例如,确定噪音分子标记的数量可包括:将信号负二项分布(两个负二项分布之一)拟合至具有与计数的测序数据中的靶相关联的不同序列的分子标记的数量,其中该信号负二项分布对应于作为信号分子标记的、具有与计数的测序数据中的靶相关联的不同序列的分子标记的数量。确定噪音分子标记的数量可包括:将噪音负二项分布(两个负二项分布中的另一个)拟合至具有与计数的测序数据中的靶相关联的不同序列的分子标记的数量,其中该噪音负二项分布对应于作为噪音分子标记的、具有与计数的测序数据中的靶相关联的不同序列的分子标记的数量。确定噪音分子标记的数量可包括使用拟合的信号负二项分布和拟合的噪音负二项分布来确定噪音分子标记的数量。
在一些实施例中,使用拟合的信号负二项分布和拟合的噪音负二项分布来确定噪音分子标记的数量包括,对于与测序数据中的靶相关联的不同序列的每个:确定该不同序列在该信号负二项分布中的信号概率。并且可以确定该不同序列在该噪音负二项分布中的噪音概率。此外,如果该信号概率小于该噪音概率,可以确定该不同序列为噪音分子标记。在一些实施例中,如果发现少于两个峰(因为需要两个峰来确定信号负二项分布和噪音负二项分布),在框644处调整分子标记计数可包括去除单元素(例如,单个碱基取代)。
在框648处,在基于邻位和基于分布的错误校正之后可以估计靶的数量以产生输出。在决策框612处,如果测序数据中的靶的测序状态是饱和的测序状态,则方法600可以前进到框648处以产生输出,而不基于定向邻接和基于分布的错误校正来调整分子标记。例如,确定的噪音分子标记的数量可以是零。
在决策框624处,如果测序数据中的靶的测序状态是测序不足状态,则方法600可以前进到框648处以产生输出,而不基于基于分布的错误校正来调整分子标记。例如,确定的噪音分子标记的数量可以是零。所述方法600在框652处结束。
免疫受体条形码错误校正
在执行测序和分析(诸如免疫受体测序和分析)时,可能会发生取代错误、引物交叉错误和pcr嵌合体错误。例如,当确定对免疫受体(诸如t细胞受体)进行编码的rna分子的出现或拷贝数量时,可能发生错误。免疫受体包括高度多样化的紧密相关的基因。因此,当与其他基因进行比较时,执行免疫受体测序和分析时测序数据中错误的可能性会更高。所述错误通常导致免疫组库多样性的过高计量。减轻这些错误的方法在本文中被称为免疫受体条形码错误校正。在一些实施例中,免疫受体条形码错误校正利用递归取代错误校正来校正分子标记和核苷酸序列中的取代错误(例如,互补决定区3(cdr3)中的取代错误)。对于给定的样品标记或细胞标记,许多不同的cdr3可以与相同的分子标记序列相关联,从而导致对免疫受体多样性的过高估计。本文披露的校正方法可以校正在分子标记和样品标记之前交叉的pcr嵌合体,然后鉴定和去除错误分子标记。
本文披露的内容包括用于确定靶的出现的方法。在一些实施例中,所述方法包括:(a)使用多个条形码(例如,随机条形码)使多个靶条形码化(例如,随机条形码化)以创建多个经条形码化的靶(例如,经随机条形码化的靶),其中,所述多个条形码中的每一个包括细胞标记和分子标记,其中,所述多个条形码中的至少两个条形码的分子标记包括不同的分子标记序列,并且其中所述多个条形码中的至少两个条形码包括具有相同的细胞标记序列的细胞标记;(b)获得所述经条形码化的靶的测序数据;以及(c)针对所述多个靶中的至少一个靶:(i)鉴定所述测序数据中的所述靶的推定序列;(ii)对与(i)中鉴定的所述测序数据中的所述靶的推定序列相关联的分子标记序列的出现进行计数;(iii)鉴定所述靶的推定序列的簇;(iv)使用(iii)中鉴定的所述靶的推定序列的簇对获得的测序数据进行折叠;(v)鉴定与所述靶的推定序列相关联的所述分子标记序列的簇;(vi)使用(v)中鉴定的所述分子标记序列的簇对所述测序数据进行折叠;(vii)鉴定组合序列的簇,其中,每个组合序列包括所述靶的序列中的序列以及所述分子标记序列中的相关联的分子标记序列;(viii)使用(vii)中鉴定的所述组合序列的簇对所述测序数据进行折叠;(ix)鉴定与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列,其中,与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列的出现小于不与所述靶的一个或多个嵌合序列相对应的所述靶的剩余一个或多个推定序列的出现;(x)从所述测序数据中去除(ix)中鉴定的与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列;以及(xi)估计所述靶的出现,其中,在(iv)、(vi)和(viii)对所述测序数据进行折叠并且在(x)中去除与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列后,估计的所述靶的出现与(ii)中计数的分子标记序列的数量相关。
本文披露的是用于确定靶的出现的方法。在一些实施例中,所述方法包括:(a)接收多个靶的测序数据,其中,所述测序数据包括所述多个靶中的靶的推定序列以及与所述测序数据中的所述靶的序列相关联的分子标记序列的出现;(b)对所述靶的推定序列进行折叠;(c)对与所述靶的推定序列相关联的分子标记序列进行折叠;以及(d)估计所述靶的出现,其中,在(b)中对所述靶的推定序列的出现进行折叠并且对在(c)中确定的噪音分子标记序列的出现进行折叠之后,估计的所述靶的出现与跟所述测序数据中的所述靶的推定序列相关联的分子标记序列的出现相关。
互补决定区(cdr)是由b细胞生成的免疫球蛋白(抗体)和t细胞生成的t细胞受体中的可变链的一部分。在抗原受体或免疫受体的可变结构域的氨基酸序列上非连续地排列有三个cdr(cdr1、cdr2和cdr3)。由于抗原受体通常由两个可变结构域组成(在两条不同的多肽链:重链和轻链上),因此,对于每对重链和轻链,总共有六个cdr可以与抗原共同接触。由于在cdr中发现了与免疫球蛋白和t细胞受体相关联的大多数序列变异,因此无法轻易地区分由在测序期间的一个或多个错误引起的对cdr进行编码的核苷酸序列中的变异和在对cdr进行编码的核苷酸序列中存在的变异。在可变结构域内,在多肽链的可变(v)区发现cdr1和cdr2,而cdr3包括v区的一部分以及重链的所有多样性(d)区和连接(j)区。轻链包含v区和j区,而不包含d区。cdr3是cdr中变化最大的。
图7是示出了基于递归取代错误校正(rsec,在本文中也称为定向邻接)的免疫受体条形码校正的非限制性示例性实施例的示意图。包括细胞标记和分子标记的条形码(例如,随机条形码)可以用于确定对免疫受体进行编码的mrna分子的序列、或通常用于确定目的靶。cdr3包括v区的一部分以及d区和j区的全部。如所展示的,样品制备和测序期间的取代错误可能在d区和j区发生(用“*”指示)。尽管未展示,但是取代错误也可能在v区中发生。另外,分子标记(ml,用“*”指示)中可能发生测序错误。递归取代错误校正(rsec)可以用于校正此类错误。例如,rsec可以用于首先调整cdr3序列的计数。随后,rsec可以用于调整分子标记的计数。可选地,通过将测序数据中的每个cdr3序列和相关联的分子标记视为一个序列,rsec可以用于同时调整cdr3序列和分子标记的计数。在一些实施例中,可以首先使用rsec调整分子标记的计数。
图8是示出了校正核苷酸序列和分子标记中的错误(基于递归取代错误校正)以及校正归因于一个或多个pcr嵌合体的测序数据中的错误的非限制性示例性实施例800的流程图。在接受多个经条形码化的靶(例如,经随机条形码化的靶)的测序数据后,在框804处开始方法800。在一些实施例中,方法800进一步包括使用多个条形码(例如,随机条形码)使多个靶条形码化(例如,随机条形码化)以创建多个经条形码化的靶,其中,所述多个条形码中的每一个包括分子标记和/或细胞标记。在一些实施例中,方法800进一步包括对多个经条形码化的靶进行测序以获得测序数据。所述多个靶可以包括细胞以下项的靶:整个转录组、基因(例如,对免疫受体(诸如t细胞受体)进行编码的基因)、可变序列(例如,对免疫受体进行编码的可变(v)区、多样性(d)区、连接(j)区)或其任何组合。
在框808处,方法800可以包括:对于所述多个靶中的一个或多个:对具有与测序数据中的靶相关联的不同序列的分子标记的数量进行确定(例如计数)。在一些实施例中,对具有与测序数据中的靶相关联的不同序列的分子标记的数量进行计数可以包括:鉴定测序数据中的所述靶的推定序列(例如,免疫受体序列,诸如cdr3序列);以及对与鉴定的所述测序数据中的所述靶的推定序列相关联的分子标记序列的出现进行计数。
靶(例如,cdr3)的推定序列可以彼此相差一个或多个核苷酸。在仅一个序列是真实序列或正确序列的意义上,所述序列是推定的(例如,每个细胞仅存在一个正确的cdr3序列)。所述靶的推定序列可以彼此相差至少一个核苷酸。
在框812处,方法800可以包括基于递归取代错误校正(在本文中也称为定向邻接)调整目的靶的推定核苷酸序列的计数。在一些实施例中,基于rsec调整核苷酸序列计数可以类似于基于参照图5描述的定向邻接调整分子标记计数。例如,基于定向邻接调整核苷酸序列计数可以包括:鉴定所述测序数据中的所述靶的推定序列;对与鉴定的所述测序数据中的所述靶的推定序列相关联的分子标记序列的出现进行计数;鉴定所述靶的推定序列的簇;以及使用鉴定的所述靶的推定序列的簇对获得的测序数据进行折叠。鉴定所述靶的推定序列的簇可以包括使用rsec鉴定所述靶的推定序列的簇。使用鉴定的所述靶的推定序列的簇对获得的测序数据进行折叠可以包括:将所述一个或多个子序列中的子序列的出现归因于所述子序列的亲本序列。
在一些实施例中,簇内所述靶的推定序列可以在彼此的第一预定定向邻接阈值内。在不同的实施方式中,第一预定定向邻接阈值可以是不同的。在一些实施例中,第一定向邻接阈值可以是汉明距离、或约1、2、3、4、5、6、7、8、9、10、或在这些值的任何两个之间的数字或范围。在一些实施例中,第一定向邻接阈值可以是汉明距离,至少或至多1、2、3、4、5、6、7、8、9或10。簇内所述靶的推定序列可以包括一个或多个亲本序列以及所述一个或多个亲本序列的一个或多个子序列。亲本序列的出现的数量可以大于或等于第一预定定向邻接出现阈值。在不同的实施方式中,第一预定定向邻接出现阈值可以是不同的。在一些实施例中,所述第一预定定向邻接出现阈值可以是小于一的子序列的出现的数量的两倍。在一些实施例中,第一预定定向邻接出现阈值可以是、或约子序列的出现的1.5倍、2倍、3倍、4倍、5倍、6倍、7倍、8倍、9倍、10倍、或在这些值的任何两个之间的数字或范围。在一些实施例中,第一预定定向邻接出现阈值可以是子序列的出现的至少或至多1.5倍、2倍、3倍、4倍、5倍、6倍、7倍、8倍、9倍、或10倍。
在框816处,方法800可以包括基于rsec调整分子标记的计数。在一些实施例中,基于定向邻接调整分子标记计数可以参考图5描述。例如,基于定向邻接调整分子标记计数可以包括:鉴定与所述靶的推定序列相关联的所述分子标记序列的簇;以及使用鉴定的所述分子标记序列的簇对所述测序数据进行折叠。鉴定与所述靶的推定序列相关联的所述分子标记序列的簇可以包括使用定向邻接鉴定与所述靶的推定序列相关联的所述分子标记序列的簇。簇内所述靶的分子标记序列可以在彼此的第二预定定向邻接阈值内。在不同的实施方式中,第二预定定向邻接阈值可以是不同的。在一些实施例中,第二定向邻接阈值可以是汉明距离、或约1、2、3、4、5、6、7、8、9、10、或在这些值的任何两个之间的数字或范围。在一些实施例中,第二定向邻接阈值可以是汉明距离,至少或至多1、2、3、4、5、6、7、8、9或10。簇内所述靶的推定分子标记序列可以包括一个或多个亲本分子标记序列以及所述一个或多个亲本分子标记序列的一个或多个子分子标记序列。亲本分子标记序列的出现可以大于或等于第二预定定向邻接出现阈值。在不同的实施方式中,第二预定定向邻接出现阈值可以是不同的。在一些实施例中,所述第二预定定向邻接出现阈值是小于一的子分子标记序列的出现的两倍。在一些实施例中,第二预定定向邻接出现阈值可以是、或约子序列的出现的1.5倍、2倍、3倍、4倍、5倍、6倍、7倍、8倍、9倍、10倍、或在这些值的任何两个之间的数字或范围。在一些实施例中,第二预定定向邻接出现阈值可以是子序列的出现的至少或至多1.5倍、2倍、3倍、4倍、5倍、6倍、7倍、8倍、9倍、或10倍。使用鉴定的与所述靶的序列相关联的所述分子标记序列的簇对所述测序数据进行折叠可以包括:将所述一个或多个子分子标记序列中的子分子标记序列的出现归因于所述子分子标记序列的亲本分子标记。
在框820处,方法800可以包括任选地基于rsec同时调整核苷酸序列和分子标记的计数。基于定向邻接同时调整核苷酸序列和分子标记的计数可以包括鉴定组合序列的簇,其中,每个组合序列包括所述靶的序列中的序列以及所述分子标记序列中的相关联的分子标记序列;以及使用鉴定的所述组合序列的簇对所述测序数据进行折叠。鉴定所述组合序列的簇可以包括使用定向邻接来鉴定所述组合序列的簇。簇内的组合序列可以在彼此的第三预定定向邻接阈值内。在一些实施例中,第三定向邻接阈值可以是汉明距离、或约1、2、3、4、5、6、7、8、9、10、或在这些值的任何两个之间的数字或范围。在一些实施例中,第三定向邻接阈值可以是汉明距离,至少或至多1、2、3、4、5、6、7、8、9或10。所述簇内的所述组合序列可以包括一个或多个亲本组合序列以及所述一个或多个亲本组合序列的一个或多个子组合序列,并且其中,所述亲本组合序列的出现大于或等于第三预定定向邻接出现阈值。在不同的实施方式中,第三预定定向邻接出现阈值可以是不同的。在一些实施例中,所述第三预定定向邻接出现阈值是小于一的子分子标记序列的出现的两倍。在一些实施例中,第三预定定向邻接出现阈值可以是、或约子序列的出现的1.5倍、2倍、3倍、4倍、5倍、6倍、7倍、8倍、9倍、10倍、或在这些值的任何两个之间的数字或范围。在一些实施例中,第三预定定向邻接出现阈值可以是子序列的出现的至少或至多1.5倍、2倍、3倍、4倍、5倍、6倍、7倍、8倍、9倍、或10倍。使用(vii)中鉴定的所述组合序列的簇对所述测序数据进行折叠可以包括:将所述一个或多个子组合序列中的子组合序列的出现归因于所述子组合序列的亲本组合序列。
在框824处,可以任选地确定测序数据中的靶的测序状态。测序数据中的靶的测序状态可包括、或是测序不足。在决策框828处,可以任选地确定测序数据中的靶的测序状态是否是测序不足状态。例如,如果靶的深度(例如平均、最小、或最大深度)小于、或小于约1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100、或在这些的任何两个之间的数字或范围,则可以认为所述靶具有测序不足状态。作为另一个实例,如果靶的深度小于至少、或至多1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、或100,则可以认为所述靶具有测序不足状态。
在一些实施例中,测序不足状态可以通过具有小于预定的测序不足阈值的深度(例如,平均、最小、或最大深度)的靶来确定。在不同实现方式中,测序不足阈值可以是不同的。例如,测序不足阈值可以是、或是约1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100、或或在这些值的任何两个之间的数字或范围。作为另一个实例,测序不足阈值可以是至少、或至多1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、或100。
在一些实施例中,测序不足状态可以取决于条形码(例如,随机条形码)的具有不同序列的分子标记的数量。例如,如果条形码包括、或约1000、2000、3000、4000、5000、6000、6561、7000、8000、9000、10000、20000、30000、40000、50000、60000、65532、70000、80000、90000、100000个具有不同序列的分子标记、或在这些值的任何两个之间的数字或范围的具有不同序列的分子标记,则测序不足阈值可以是10(或另一个阈值数量)。作为另一个实例,如果条形码包括至少、或至多1000、2000、3000、4000、5000、6000、6561、7000、8000、9000、10000、20000、30000、40000、50000、60000、65532、70000、80000、90000、或100000,则测序不足阈值可以是10(或另一个阈值数量)。在一些实施例中,测序不足状态可以不取决于条形码(例如,随机条形码)的具有不同序列的分子标记的数量。
在决策框828处,如果测序数据中靶的测序状态不是测序不足状态,则方法800可以前进到决策框832,以确定在框812、816和820处进行调整后是否在核苷酸序列和/或分子标记中剩余任何单例(例如,单个碱基取代)。如果剩余至少一个单例,则方法800可以前进到框836,其中,可以去除与嵌合体相对应的分子标记计数。
在框836处,方法800可以包括去除与嵌合体相对应的分子标记计数。图9是免疫受体嵌合体(或靶嵌合体)的一种可能来源的示意图。如所展示的,许多不同的cdr3序列(或靶的推定序列)可能具有相同的ml(和样品标记或细胞标记)或与之相关联,这可能导致tcr多样性被过高估计。两个或更多个真实cdr3序列可以在pcr期间交叉。例如,在条形码化之后(例如,在图2的块224之后),两个真实cdr3序列(在图9中标记的cdr3-1和cdr3-2)可以与两个不同的分子标记(标记的ml-1和ml-2)相关联。两个(或更多个)真实cdr3序列可以是来自两个不同细胞的两个不同的cdr3序列,所述细胞不是pcr交叉导致的。免疫受体嵌合体可以在pcr期间形成(例如,在图2中的框228处),使得多个不同的cdr3序列(例如,两个不同的cdr3序列)可以具有相同的ml。例如,ml-1可以与cdr3-1以及cdr3-1和cdr3-2的嵌合体相关联。作为另一个实例,ml-2可以与cdr3-2以及cdr3-1和cdr3-2的嵌合体相关联。去除与嵌合体相对应的分子标记计数可能是有利的。
参考图8,去除与嵌合体相对应的分子标记计数可以包括鉴定与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列,其中,与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列的出现小于不与所述靶的一个或多个嵌合序列相对应的所述靶的剩余一个或多个推定序列的出现;以及在所述测序数据中去除鉴定的与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列。鉴定与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列可以包括:鉴定与所述多个分子序列中的一个分子标记序列相关联的所述靶的推定序列;以及鉴定与所述一个分子标记序列相关联的所述靶的推定序列中的推定序列,所述分子标记序列的出现小于与所述靶的一个或多个嵌合序列中的嵌合序列相对应的嵌合出现阈值。在不同的实施方式中,嵌合出现阈值可以是不同的。在一些实施例中,嵌合出现阈值的值可以是与一个分子标记序列相关联的所述靶的推定序列中的推定序列的出现,所述出现大于所述靶的推定序列中的任何其他序列的出现。在一些实施例中,嵌合出现阈值可以是与一个分子标记序列相关联的所述靶的推定序列中的推定序列的出现,所述出现大于通过偏移调整(例如,减法)的所述靶的推定序列中的任何其他序列的出现。在不同的实施方式中,所述偏移可以是不同的。在一些实施例中,所述偏移可以是、或约1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100、或在这些值的任何两个之间的数字或范围。在一些实施例中,所述偏移可以是至少、或至多1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、或100。
在框812、816和820处对测序数据进行折叠之后,在框824之后估计的靶的出现与在框808处计数的分子标记序列的数量相关。在框812、816和820处对测序数据进行折叠并且在框836处去除与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列之后,在框836之后估计的靶的出现与在框808处计数的分子标记序列的数量相关。
在框840处,可以在确定具有与测序数据中的所述靶相关联的不同序列的噪音分子标记的数量之前,将伪点任选地添加到具有与测序数据中的所述靶相关联的不同序列的分子标记的数量。例如,如果具有与测序数据中的靶相关联的不同序列的分子标记的数量小于伪点阈值,则方法800可以包括在确定具有与测序数据中的靶相关联的不同序列的噪音分子标记的数量之前,将伪点添加至具有与测序数据中的靶相关联的不同序列的分子标记的数量中。在不同实现方式中,伪点阈值可以是不同的。例如,如果条形码(例如,随机条形码)包括具有不同序列的约6561个分子标记,则伪点阈值可以是、或是约1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100、或在这些值的任何两个之间的数字或范围。作为另一个实例,如果条形码(例如,随机条形码)包括具有不同序列的约6561个分子标记,则伪点测序阈值可以是至少、或至多1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、或100。在不同的实施方式中,添加的伪点可以具有不同分子标记计数。例如,伪点的分子标记计数可以是、或是约0.0001、0.001、0.01、0.1、1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100、或在这些值的任何两个之间的数字或范围。作为另一个实例,伪点的分子标记计数可以是至少、或至多0.0001、0.001、0.01、0.1、1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、或100。
在框844处,可以使用基于分布的错误校正方法来调整分子标记计数。执行基于分布的错误校正方法可包括确定具有与测序数据中的靶相关联的不同序列的噪音分子标记的数量。确定噪音分子标记的数量可包括:将两个分布(诸如两个负二项分布)拟合至具有测序数据中的与所述靶相关联的不同序列的分子标记的数量。例如,确定噪音分子标记的数量可包括:将信号负二项分布(两个负二项分布之一)拟合至具有与计数的测序数据中的靶相关联的不同序列的分子标记的数量,其中该信号负二项分布对应于作为信号分子标记的、具有与计数的测序数据中的靶相关联的不同序列的分子标记的数量。确定噪音分子标记的数量可包括:将噪音负二项分布(两个负二项分布中的另一个)拟合至具有与计数的测序数据中的靶相关联的不同序列的分子标记的数量,其中该噪音负二项分布对应于作为噪音分子标记的、具有与计数的测序数据中的靶相关联的不同序列的分子标记的数量。确定噪音分子标记的数量可包括使用拟合的信号负二项分布和拟合的噪音负二项分布来确定噪音分子标记的数量。
在一些实施例中,使用拟合的信号负二项分布和拟合的噪音负二项分布来确定噪音分子标记的数量包括,对于与测序数据中的靶相关联的不同序列的每个:确定该不同序列在该信号负二项分布中的信号概率。并且可以确定该不同序列在该噪音负二项分布中的噪音概率。此外,如果该信号概率小于该噪音概率,可以确定该不同序列为噪音分子标记。在一些实施例中,如果发现少于两个峰(因为需要两个峰来确定信号负二项分布和噪音负二项分布),在框644处调整分子标记计数可包括去除单元素(例如,单个碱基取代)。
在框812、816和820处对所述测序数据进行折叠并且在框836处去除与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列后,执行基于分布的错误校正可以包括:在框812、816和820处对所述测序数据进行折叠并且在框836处去除与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列后,将与所述靶的推定序列相关联的分子标记序列阈值化,以确定与在框808处计数的所述测序数据中的所述靶的序列相关联的信号分子标记序列和噪音分子标记序列。将与所述靶的推定序列相关联的所述分子标记序列阈值化可以包括对所述靶的所述分子标记序列进行统计分析。进行所述统计分析可以包括:将与所述靶的推定序列相关联的所述分子标记序列及其出现拟合至两个负二项分布;使用所述两个负二项分布来确定信号分子标记序列的出现n;以及在(iv)、(vi)和(viii)对所述测序数据进行折叠并且在(x)中去除与所述靶的一个或多个嵌合序列相对应的所述靶的一个或多个推定序列后,从在(b)中获得的测序数据去除所述噪音分子标记序列,其中,所述噪音分子标记序列包括其出现小于第n个最丰富分子标记的出现的分子标记序列,并且其中所述信号分子标记序列包括其出现大于或等于第n个最丰富分子标记的出现的分子标记序列。这两个负二项分布可以包括与所述信号分子标记序列相对应的第一负二项分布以及与所述噪音分子标记序列相对应的第二负二项分布。
在框848处,在基于邻位和基于分布的错误校正之后可以估计靶的数量以产生输出。在决策框828处,如果测序数据中的靶的测序状态是测序不足状态,则方法800可以前进到框848处以产生输出,而不基于基于分布的错误校正来调整分子标记。例如,噪音分子标记的数量可以是零。在决策框832处,如果经调整的测序数据中没有单例,则方法800可以前进到框848处以产生输出,而不在基于分布的错误校正的基础上来调整分子标记。所述方法800在框852处结束。
测序
在一些实施例中,估算不同的经标记的或经条形码化的靶(例如,经随机条形码化的靶)的数量可以包括确定经标记的靶、空间标记、分子标记、样品标记、细胞标记、或其任何产物(例如,经标记的扩增子、或经标记的cdna分子)的序列。扩增的靶可以经受测序。确定经条形码化的靶(例如,经随机条形码化的靶)或其任何产物的序列可以包括进行测序反应以确定样品标记、空间标记、细胞标记、分子标记的至少一部分的序列,经条形码化的靶、其互补体、其反向互补体的至少一部分的序列,或其任何组合的序列。
可以使用多种测序方法确定经条形码化的靶或经随机条形码化的靶的序列(例如,扩增的核酸、标记的核酸、标记的核酸的cdna拷贝等),这些方法包括但不限于杂交测序(sbh)、连接法测序(sbl)、量化增量荧光核苷酸附加测序(quantitativeincrementalfluorescent核苷酸additionsequencing)(qifnas)、分段连接与断裂、荧光共振能量转移(fret)、分子信标、taqman报告探针消化、焦磷酸测序、荧光原位测序(fisseq)、fisseq珠、摆动测序(wobblesequencing)、多重测序、聚合集群(polymerizedcolony)(polony)测序;纳米格滚环测序(nanogridrollingcirclesequencing,rolony)、等位基因特异性寡核苷酸连接检验(allele-specificoligoligationassay)(例如,寡核苷酸连接检验(ola)、使用连接的线性探针和滚环扩增(rca)读出、连接的持锁探针的单模板分子(singletemplatemolecule)ola、或使用连接的环形持锁探针和滚环扩增(rca)读出的单模板分子ola)等。
在一些实施例中,确定经条形码化的靶(例如,经随机条形码化的靶)或其任何产物的序列包括配对端测序、纳米孔测序、高通量测序、鸟枪法测序、染料终止剂测序、多重引物dna测序、引物步移、桑格双脱氧测序法、马克西姆-吉尔伯特(maxim-gilbert)测序、焦磷酸测序、真单分子测序或其任何组合。可替代地,可以通过电子显微镜分析法或化学敏感场效应晶体管(chemfet)阵列来确定经条形码化的靶或其任何产物的序列。
也可以使用高通量测序方法,诸如使用平台(诸如roche454、illuminasolexa、abi-solid、iontorrent、completegenomics、pacificbioscience、helicos、或polonator平台)的循环阵列测序。在一些实施例中,测序可以包括miseq测序。在一些实施例中,测序可以包括hiseq测序。
经条形码化的靶(例如,经随机条形码化的靶)可以包括代表来自生物体基因组基因的约0.01%至生物体基因组基因的约100%的核酸。例如,可以使用包括多个多聚体的靶标互补区域,通过从所述样品中捕获含有互补序列的基因,对约0.01%的生物体基因组基因至约100%的生物体基因组基因进行测序。在一些实施例中,经条形码化的靶包括代表从约0.01%的生物体转录组转录物至约100%的生物体转录组转录物的核酸。例如,可以使用包括聚(t)尾的靶标互补区域,通过从所述样品中捕获mrna,对约0.501%的生物体转录组转录物至约100%的生物体转录组转录物进行测序。
确定多个条形码(例如随机条形码)的空间标记和分子标记的序列可包括对多个条形码的0.00001%、0.0001%、0.001%、0.01%、0.1%、1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、20%、30%、40%、50%、60%、70%、80%、90%、99%、100%、或在这些值的任何两个之间的数字或范围进行测序。确定多个条形码的标记(例如样品标记、空间标记、和分子标记)的序列可包括对多个条形码中的1、10、20、30、40、50、60、70、80、90、100、103、104、105、106、107、108、109、1010、1011、1012、1013、1014、1015、1016、1017、1018、1019、1020个、或在这些值的任何两个之间的数字或范围进行测序。对多个条形码中的一些或所有进行测序可包括产生具有、具有约、具有至少、或具有至多10、20、30、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900、1000、2000、3000、4000、5000、6000、7000、8000、9000、10000个、或在这些值的任何两个之间的数字或范围内的核苷酸或碱基的读数长度的序列。
测序可以包括对经条形码化的靶(例如,经随机条形码化的靶)的至少或至少约10、20、30、40、50、60、70、80、90、100个或更多个核苷酸或碱基对进行测序。例如,测序可包括通过对多个经条形码化的靶进行聚合酶链式反应(pcr)扩增,产生测序数据,其中序列具有50、75、或100个、或更多个核苷酸的读数长度。测序可包括对经条形码化的靶的至少或至少约200、300、400、500、600、700、800、900、1,000个或更多个核苷酸或碱基对进行测序。测序可包括对经条形码化的靶的至少或至少约1500、2000、3000、4000、5000、6000、7000、8000、9000、或10000个或更多个核苷酸或碱基对进行测序。
测序可以包括至少约200、300、400、500、600、700、800、900、1,000个或更多个测序读数/运行。在一些实施例中,测序包括每次运行对至少或至少约1500、2000、3000、4000、5000、6000、7000、8000、9000、或10000个或更多个测序读数进行测序。测序可以包括小于或等于约1,600,000,000个测序读数/运行。测序可以包括小于或等于约200,000,000个读数/运行。
样品
在一些实施例中,多个靶可以包括在一个或多个样品中。样品可包括一个或多个细胞、或者来自一个或多个细胞的核酸。样品可以是单细胞或来自单细胞的核酸。一个或多个细胞可以是一种或多种细胞类型。一种或多种细胞类型中的至少一种可以是脑细胞、心脏细胞、癌细胞、循环肿瘤细胞、器官细胞、上皮细胞、转移性细胞、良性细胞、原代细胞、循环细胞或其任何组合。
用于在本披露的方法中使用的样品可以包括一个或多个细胞。样品可以指一个或多个细胞。在一些实施例中,多个细胞可包括一种或多种细胞类型。一种或多种细胞类型中的至少一种可以是脑细胞、心脏细胞、癌细胞、循环肿瘤细胞、器官细胞、上皮细胞、转移性细胞、良性细胞、原代细胞、循环细胞或其任何组合。在一些实施例中,该细胞是从癌组织切除的癌细胞,例如乳腺癌、肺癌、结肠癌、前列腺癌、卵巢癌、胰腺癌、脑癌、黑素瘤和非黑素瘤皮肤癌等。在一些实施例中,该细胞源自癌症,但是从体液收集(例如,循环肿瘤细胞)。癌症的非限制性实例可以包括腺瘤、腺癌、鳞状细胞癌、基底细胞癌、小细胞癌、大细胞未分化癌、软骨肉瘤、以及纤维肉瘤。样品可包括组织、单层细胞、固定的细胞、组织切片、或其任何组合。样品可包括生物学样品、临床样品、环境样品、生物学流体、来自受试者的组织或细胞。样品可以获得自人、哺乳动物、狗、大鼠、小鼠、鱼、蝇、蠕虫、植物、真菌、细菌、病毒、脊椎动物、或无脊椎动物。
在一些实施例中,该细胞是已经被病毒感染并含有病毒寡核苷酸的细胞。在一些实施例中,病毒感染可以由如单链(+链或“有义”)dna病毒(例如,细小病毒)、或双链rna病毒(例如,呼吸道肠道病毒)的病毒引起。在一些实施例中,所述细胞是细菌。这些可以包括革兰氏阳性细菌或革兰氏阴性细菌。在一些实施例中,所述细胞是真菌。在一些实施例中,所述细胞是原生动物或其他寄生虫。
如本文使用的,术语“细胞”可以指一个或多个细胞。在一些实施例中,细胞是正常细胞,例如,处于不同发育阶段的人细胞,或来自不同器官或组织类型的人细胞。在一些实施例中,该细胞是非人类细胞,例如其他类型的哺乳动物细胞(例如小鼠、大鼠、猪、狗、牛或马)。在一些实施例中,所述细胞是其他类型的动物或植物细胞。在其他实施例中,细胞可以是任何原核或真核细胞。
数据分析和显示软件
靶的空间分辨率的数据分析和可视化
本披露提供了使用空间标记用条形码化(例如,随机条形码化)和数字计数来估计靶的数目和位置的方法。从披露的方法获得的数据可以在图谱上可视化。可以使用用本文所述方法生成的信息来构建来自样品的靶数目和位置的图谱。图谱可用于定位靶的物理位置。图谱可用于鉴定多个靶的位置。多个靶可以是相同的靶种类,或多个靶可以是多个不同的靶。例如,可以构建脑的图谱,以显示多个靶的数字计数和位置。
可以从来自单个样品的数据生成图谱。可以使用来自多个样品的数据构建图谱,从而生成组合图谱。可以使用来自数十、数百和/或数千个样品的数据构建图谱。从多个样品构建的图谱可以示出与多个样品共同区域相关联的靶的数字计数的分布。例如,重复的测定可以显示在同一个图谱上。可以在相同的图上显示至少1、2、3、4、5、6、7、8、9、或10个,或更多个重复(例如,覆盖)。可以在相同的图上显示至多1、2、3、4、5、6、7、8、9、或10个,或更多个重复(例如,覆盖)。空间分布和靶数目可以由各种统计来表示。
组合来自多个样品的数据可以增加组合图谱的位置分辨率。多个样品的方向可以通过常见的界标来记录,其中跨样品的各个位置测量是至少部分不连续的。一个特别的例子是在一个轴上使用超薄切片机对样品进行切片,并且然后沿着不同的通路对第二个样品进行切片。组合数据集将给出与靶标的数字计数相关联的三维空间位置。复用上述方法将允许数字计数统计的高分辨率三维图谱。
在所述仪器系统的一些实施例中,所述系统将包括计算机可读介质,其包括用于为通过进行单细胞、条形码化测定(例如,随机条形码化测定)而生成的序列数据集提供数据分析的代码。可以由数据分析软件提供的数据分析功能的实例包括但不限于(i)用于解码/多路解编通过对在运行测定中产生的条形码文库(例如,随机条形码文库)进行测序所提供的样品标记、细胞标记、空间标记和分子标记以及靶序列数据的算法,(ii)用于基于数据确定每个细胞每个基因的读数数量和每个细胞每个基因的独特转录物分子数量、并且创建汇总表的算法,(iii)对序列数据的统计分析,例如用于通过基因表达数据对细胞聚类,或用于预测确定每个细胞每个基因的转录物分子数目的置信区间等,(iv)用于鉴定罕见细胞亚群的算法,例如使用主成分分析、分层聚类、k均值聚类、自组织映射、神经网络等,(v)基因序列数据与已知参考序列比对以及突变、多态性标记物和剪接变体的检测的序列比对能力,以及(vi)分子标记的自动聚类,以补偿扩增或测序错误。在一些实施例中,可商购的软件可用于执行数据分析的全部或一部分,例如,可以使用七桥(sevenbridges,https://www.sbgenomics.com/)软件来编译在整个细胞集合中每个细胞中出现的一个或多个基因的拷贝数目表。在一些实施例中,数据分析软件可以包括用于以有用的图形格式输出测序结果的选项,例如,指示在细胞集合的每个细胞中存在的一个或多个基因的拷贝数的热图。在一些实施例中,数据分析软件还可以包括用于从测序结果提取生物学意义的算法,例如通过将在细胞集合的每个细胞中存在的一个或多个基因的拷贝数与一种细胞、一种稀有细胞、或来源于具有特定疾病或病症的受试者的细胞相关联。在一些实施例中,数据分析软件还可以包括用于比较跨不同生物样品的细胞群的算法。
在一些实施例中,所有数据分析功能都可以打包在单个软件包中。在一些实施例中,完整的数据分析能力集合可以包括一套软件包。在一些实施例中,数据分析软件可以是独立于测定仪器系统的使得使用者可用的独立包。在一些实施例中,软件可以是基于网络的,并且可以允许用户共享数据。
在一些实施例中,所有数据分析功能都可以打包在单个软件包中。在一些实施例中,完整的数据分析能力集合可以包括一套软件包。在一些实施例中,数据分析软件可以是独立于测定仪器系统的使得使用者可用的独立包。在一些实施例中,软件可以是基于网络的,并且可以允许用户共享数据。
系统处理器和网络
通常,适用于当前披露的仪器系统(如图10所说明的)的方法中使用的计算机或处理器可以被进一步理解为逻辑设备,其可以读取来自介质1011或网络端口1005的指令,其可以任选地与具有固定介质1012的服务器1009连接。系统1000(如图10所示的)可以包括cpu1001、磁盘驱动器1003、任选的输入装置(如键盘1015或鼠标1016)以及任选的监测器1007。可以通过从指定的通信媒质传至在本地或远程位置的服务器来实现数据通信。通信媒质可以包括发送或接收数据的任何工具。例如,通信介质可以是网络连接、无线连接或因特网连接。这样的连接可以提供经由万维网的通信。可以设想的是,与本披露有关的数据可以通过这样的网络或连接进行传输,以由接收方1022接收或审阅,如图10所说明的。
图11说明了可以结合本披露的实例实施例使用的计算机系统1100的第一实例架构的示例性实施例。如图11所描绘的,该示例性计算机系统可以包括用于处理指令的处理器1102。处理器的非限制性实例包括:英特尔xeontm处理器、amdopterontm处理器、三星32位riscarm1176jz(f)-sv1.0tm处理器、armcortex-a8三星s5pc100tm处理器、armcortex-a8苹果a4tm处理器、迈威尔(marvell)pxa930tm处理器或功能等效的处理器。多个执行线程可以用于并行处理。在一些实施例中,也可以使用多处理器或具有多个核心的处理器,无论是以单个计算机系统、成群地还是通过网络跨系统分布的,所述网络包括多个计算机、手机、或个人数据助理装置。
如图11所说明的,可以将高速缓存1104连接至或并入处理器1102中,以便为最近已经被或频繁地被处理器1102使用的指令或数据提供高速存储器。通过处理器总线1108将处理器1102与北桥1106连接。北桥1106通过存储总线1112与随机存取存储器(ram)1110连接并且管理处理器1102对ram1110的访问。北桥1106还通过芯片集总线1116与南桥1114连接。南桥1114进而与外设总线1118连接。外设总线可以是例如pci、pci-x、pciexpress或其他外设总线。北桥和南桥通常被称为处理器芯片集并且管理处理器、ram与外设总线1118上的外设部件之间的数据传输。在一些替代性架构中,可以将北桥的功能性并入处理器中而取代使用单独的北桥芯片。
在一些实施例中,系统1100可以包括附接至外设总线1118的加速器卡1122。加速器可以包括现场可编程门阵列(fpga)或其他用于加速某些处理的硬件。例如,加速器可以用于自适应性数据重构或用于评价扩展集处理中所用的代数表达式。
软件和数据被存储在外部存储器1124中并且可以被加载到ram1110或缓存1104中,用于为处理器所用。系统1100包括用于管理系统资源的操作系统;操作系统的非限制性实例包括:linux、windowstm、macostm、blackberryostm、iostm、和其他功能等效的操作系统,以及在操作系统之上运行的用于根据本发明的实例实施例管理数据存储和优化的应用软件。
在这个实例中,系统1100还包括与外设总线连接的网络接口卡(nic)1120和1121,用于为外部存储器(如附网存储(nas))和其他可以用于分布式并行处理的计算机系统提供网络接口。
图12展示了示出网络1200的示例性图表,所述网络具有适用于本披露的方法的多个计算机系统1202a和1202b、多个手机和个人数据助理1202c、以及附网存储(nas)1204a和1204b。在实例实施例中,系统1212a、1212b和1212c可以管理数据存储并针对存储在附网存储(nas)1214a和1214b中的数据优化数据访问。数学模型可以用于数据并且使用跨计算机系统1212a和1212b、和手机以及个人数据助理系统1212c的分配式并行处理进行评价。计算机系统1212a和1212b和手机以及个人数据助理系统1212c还可以为存储在附网存储(nas)1214a和1214b中的数据的自适应性数据重构提供并行处理。图12仅说明了一个实例,并且多种多样的其他计算机架构和系统也可以与本发明的不同实施例结合使用。例如,刀片式服务器可以用来提供并行处理。处理器刀片可以通过底板进行连接,以提供并行处理。存储器也可以通过单独的网络接口与底板附接或作为附网存储(nas)。
在一些实例实施例中,处理器可以保持分开的存储空间并且通过网络接口、底板或其他连接器传输数据,用于通过其他处理器进行并行处理。在其他实施例中,一些或所有处理器可以使用共享的虚拟地址存储空间。
图13显示了根据一个实例实施例的多处理器计算机系统1300的示例性框图,所述系统使用共享的虚拟地址存储空间。所述系统包括可以访问共享的存储子系统1304的多个处理器1302a-f。该系统在存储子系统1304中并入了多个可程编硬件存储算法处理器(map)1306a-f。map1306a-f各自可以包括存储器1308a-f以及一个或多个现场可编程门阵列(fpga)1310a-f。map提供了可配置功能单元并且具体算法或算法部分可以被提供给fpga1310a-f,用于与相应的处理器密切配合进行处理。例如,map可以用于相对于数据模型来评价代数表达式以及用于在实例实施例中进行自适应性数据重构。在这个实例中,每个map都可被所有处理器全球访问,用于这些目的。在一种配置中,每个map都可以使用直接内存访问(dma)来访问相关联的存储器1308a-f,从而允许它独立于相应的微处理器1302a-f并且与其不同步地执行任务。在这种配置中,map可以将结果直接给至另一map,用于流水操作和并行执行算法。
以上计算机架构和系统仅仅是实例,并且多种多样的其他计算机、手机和个人数据助理架构和系统可以与实例实施例结合使用,包括使用一般处理器、协同处理器、fpga和其他可编程逻辑装置、片上系统(soc)、专用集成电路(asic)以及其他处理和逻辑元件的任何组合的系统。在一些实施例中,全部或部分的计算机系统可以在软件或硬件中实施。任何种类的数据存储介质都可以与实例实施例结合使用,包括随机存取存储器、硬盘驱动器、闪速存储器、磁带驱动器、磁盘阵列、附网存储(nas)以及其他局部或分布式数据存储装置和系统。
在实例实施例中,可以使用在任何以上或其他计算机架构和系统上执行的软件模块来实施本披露的计算机子系统。在其他实施例中,可以部分地或完全地在以下项中实现该系统的功能:固件、可编程逻辑装置(如现场可编程门阵列(fpga))、片上系统(sol)、专用集成电路(asic)或其他处理和逻辑元件。例如,可以通过使用硬件加速器卡(如加速器卡)通过硬件加速来实施集处理器和优化器。
实例
以上讨论的实施例的一些方面在以下实例中进一步详细披露,其不是旨在以任何方式限制本披露的范围。
实例1
板中每个ml对于高表达基因-actb的ml覆盖
此实例证明了在测序或pcr期间衍生的ml错误的不同分布通常具有与真ml不同的分布。
除绝对基因表达计数和pcr偏差校正外,ml还可以提供对文库制备程序和测序数据的统计质量更好地理解。当查看呈现相同基因ml的读数的数量(称为ml覆盖)时,可以检测在文库制备期间产生的测序错误碱基调用或pcr错误。例如,与来自仅由单个读数表示的给定sl的基因ml相比,来自由多个读数表示的给定sl的基因ml可能是精确的测量。在同一文库中存在高ml覆盖条形码的情况下,低ml覆盖条形码通常是在文库制备期间在测序运行或pcr步骤期间产生的伪影或错误。在测序或pcr期间衍生的ml错误与真ml通常具有不同分布。图15是示出了高表达基因-atcb的微孔板的每个分子标记的分子标记覆盖的示例性图,其中在错误分子标记和真实分子标记之间观察到不同的分布。图16是示出了对于高表达基因-atcb的微孔板的每个分子标记的分子标记覆盖拟合两个负二项分布的示例性图。两个负二项分布的拟合证明具有较低的分子标记深度的分子标记错误和具有较高的分子标记深度的真分子标记可以在统计学上区分。x轴是分子深度。
总之,这些数据证明在测序或pcr期间衍生的ml错误与真ml通常具有不同分布。
实例2
校正由于pcr或测序取代错误的分子标记
此实例证明了用于校正由于pcr和测序取代错误的分子标记的方法,该方法可以应用至整个转录组测定而不需要均匀覆盖的假设且不需要对完全测序状态的高测序覆盖。
在每个读数的第一个映射坐标和独特分子标记(umi)上进行重复数据删除,并且在相同的起始坐标、uml和链的情况下,读数被假定是相同的。重复数据删除后,保留了每个簇计数最高的uml(表13)。
基于每基因偏差校正分子标记(ml)。对于每个基因,用定向邻接鉴定ml的簇。如果ml在1汉明距离内且亲本ml计数≥2*(子mi计数)-1,则定向邻接方法聚簇ml。相同簇中的所有ml被认为源自相同的亲本ml,并且子ml计数被折叠到亲本ml。图17示出了分子标记校正,其中为1的成对汉明距离被过度代表。在分子标记校正后,将相距一汉明距离的分子标记聚簇并折叠为相同的亲本分子标记。图18显示了ml的校正的数量相比于读数覆盖的数量的曲线。由于保留了所有读数,可以使用此方法来去除一个碱基pcr或测序错误。
表13.在对分子标记进行重复数据删除后,在给定整个转录组测定的情况下,仅将不足数量的独特分子标记视为错误
总之,这些数据证明了校正方法,该校正方法可以用于校正或调整整个转录组测定的数据,因为所有的读数都保留。
实例3
对于高输入样品的分子标记计数
此实例描述了用作输入分子增加的独特分子标记。
当以小样品输入(如单细胞)使用时,bdprecisetm靶向测定可以是最合适允许随机和独特标记mrna的。因为在高rna/细胞输入实验中转录物的数量相对于条形码池增加,将ml的百分比再循环以标记相同基因增加并且理论上使用泊松分布进行计算(图14)。在这些情况下,在没有统计学校正的情况下,使用ml定量基因表达将在没有任何泊松校正或基于两个负二项分布的校正的情况下低估最初存在的分子数量。
在极高输入样品中,其中每个基因的mrna数量超过6561条码的整个集合,泊松校正或基于两个负二项分布的校正不再可能。例如,无论65000个或100000个输入分子,在任何情况下预计最多6561个饱和条形码。因此,看似具有高样品输入的基因和样品可以被改变,从而可能低估ml计数。
总之,这些数据证明了当使用ml定量基因表达时,需要调整原始数据。
实例4
递归取代错误校正(rsec)
此实例证明了递归取代错误校正。
在bdprecisetm靶向测定分析管道中可以应用两种协作方法以去除ml错误。简而言之,使用递归取代错误校正(rsec),鉴定源自测序碱基调用取代错误的ml错误,并调整为真ml条形码。随后,使用基于分布的错误校正(dbec)调整源自文库制备步骤或测序碱基缺失错误的ml错误。
rsec算法可以调整源自pcr或测序取代的ml错误。当检查ml覆盖时,已经观察到这些罕见的错误事件。例如,错误ml的ml覆盖可以显著地低于充分测序样品中的真ml(图15);在最初的molecularindexingtm(逆转录)步骤期间使用两个非常相似的ml的情况下,它们通常具有相似的ml覆盖并且不需要被消除。随着测序深度的增加,出现更多的ml错误,因此rsec对于调整高度测序的条形码文库的ml计数至关重要。
简而言之,rsec考虑了错误校正中的两个因素:1)ml序列的相似性;以及2)及其ml覆盖。对于每个靶基因,当它们的ml序列彼此在1个碱基(汉明距离=1)内时,ml被连接。对于在mlx与y之间的每个连接,如果:
覆盖(y)>2*覆盖(x)+1,方程(5)
其中y表示“亲本ml”,且x表示“子ml”。
基于此分配,子ml可以折叠至其亲本ml。这个过程是递归的,直至基因没有更多可鉴定的亲本/子ml。
图19显示了以上概述的递归取代错误校正的实例的示意图。在rsec校正之前,原始数据中的ml包括九个独特ml:gtcaaatt、gtcaaaat、gtcaaaaa、ttcaaaaa、ttcagaaa、ctcaaaaa、ttcaaact、ttcaaaat和ttcaaaca(seqidno:3-11)。通过应用rsec,gtcaaatt(seqidno:3)可以被折叠成gtcaaaat(seqidno:4),因为两个ml具有一个核苷酸(下划线)差异,并且mlgtcaaatt(seqidno:3)的ml计数低于gtcaaaat(seqidno:4)。进而,mlgtcaaaat(seqidno:4)可以被折叠成mlgtcaaaaa(seqidno:5)(ml序列中的差异加下划线),其具有比gtcaaaat(seqidno:4)更高的ml计数。相似地,mlttcagaaa(seqidno:7)和ctcaaaaa(seqidno:8)可以被折叠成mlttcaaaaa(seqidno:6)。mlttcaaact(seqidno:9)可以被折叠成mlttcaaaat(seqidno:10),其进而可以被折叠成mlttcaaaaa(seqidno:6)。mlttcaaaca(seqidno:11)与所有其他ml相比具有多于一个核苷酸差异,并且因此不会被折叠成其他八个ml中的任何一个。在rsec校正之前,原始ml计数是九。在rsec校正之后,ml计数是二:mlttcaaaaa(seqidno:6)和ttcaaaca(seqidno:11)。
总之,这些数据证明了使用rsec来校正原始ml计数。
实例5
ml覆盖计算
此实例描述了ml覆盖计算。
在rsec之后,计算每孔的基因ml技术以确定它们是否适合进一步校正。具有低ml覆盖(每ml<4读数)的基因绕过后续校正步骤并且在最终ml数据表中报告并且在生物信息学管道中记录为“低深度”。对于具有极高输入的基因,其中观察到可能的6561个条形码中的至少6557个,其中由于条形码多样性而使确定分子数量变得具有挑战性并且基因被标记为“饱和”。对于不符合2个决策点中的任何一个的基因ml,将前进到后续的dbec算法,并在输出日志文件中标记为“忽略(pass)”。此外,高于平均每孔650个ml的基因被记录为“高输入”,因为>5%的这些ml基于泊松分布被再循环(图15)。
总之,此实例描述了ml覆盖计算。
实例6
基于分布的错误校正(dbec)
此实例描述了基于分布的错误校正。
与rsec不同,dbec算法是区分ml是错误还是真信号的方法,而不管其ml序列如何。虽然rsec可以使用ml序列和ml覆盖信息两者来校正错误,但dbec主要依靠ml覆盖仅来校正非取代错误。如前所述,错误条形码通常具有低ml覆盖范围,这与真条形码ml覆盖范围不同;可以在ml覆盖的直方图中观察到ml覆盖的这种差异作为不同的分布(图15)。鉴于这种差异,dbec拟合两个负二项分布以统计学上区分ml错误(具有较低ml覆盖)和具有较高ml覆盖的真信号。
去除再循环的ml以实现最佳分布拟合
对于给定的基因,当检测到的ml增加时,再循环的ml的百分比(即,相同的ml用于标记来自相同基因的2个或更多个mrna)增加并且可以估计。使用泊松分布(λ非独特),从ml再循环率方程(方程(6))估计对于孔i(n非独特,i)的再循环ml的数量。如果估计的再循环的ml大于孔i中给定基因的总ml的5%,则孔i中的该基因被标记为“高输入”。对于这些“高输入”数据,靠前的ml覆盖ml将从分布拟合中消除-但保留用于以后的计数步骤-以获得更好的负二项分布。
p(x>1|λ非独特),λ非独特
=ml的数量/6561。方程(6)
对于低表达基因添加伪点
如果ml的独特数量小于10,则由于数据的稀疏性,对于拟合分布通常更具挑战性。为了缓解此问题,dbec在1%信号计数处添加伪点用于辅助分布拟合,但不影响数据。
参数估计
为了拟合两个负二项分布以将错误与信号ml分离,用于参数估计的两组起始值近似化。假设错误分布为具有平均值和离散度为1的负二项。
错误/信号概率估计
假设信号和错误分布分别为负二项(μ信号,尺寸信号)和负二项(μ错误,尺寸错误)。为了确定信号ml的数量(以升序),计算来自给定ml的读数是来自信号和错误分布的概率,直到满足方程(8),其中所有在先前ml被认为是错误ml。
p(x=r|μ=μ错误,尺寸=尺寸错误)
<p(xr|μ=μ信号,尺寸=尺寸信号)。方程(8)
总之,此实例显示了用于进行基于分布的错误校正的计算。
实例7
基于dbec校正pcr和测序错误
此实例证明了基于两个负二项分布校正pcr和测序错误。
图20a至20c示出了基于cd69的两个负二项分布校正pcr和测序错误的示例性结果。图20a示出了cd69的两个负二项分布(dn为噪音负二项分布,并且ds为信号二项分布)在图20b中的ml深度直方图中示出的ml计数数据上的拟合。图20b中的虚线示出了通过图20a中示出的两个负二项分布确定的ml信号和sl错误的分离。图20c中的垂直线示出了如基于读数的累积总和图确定的二阶导数的局部最大值。类似于图20a至20c,图21a至21c示出了基于cd3e的两个负二项分布校正pcr和测序错误的示例性结果。
总之,这些数据显示了dbec可以用于校正pcr和测序错误。
实例8
使用两个负二项分布的ml计数校正
此实例证明了使用两个负二项分布校正的十个靶的ml计数。
图22a至22j示出了使用两个负二项分布校正的数据集的非限制性示例性验证。如图22a至22j所示,校正了10个靶的ml计数。图22a至22j的每个图中的垂直线显示了使用两个负二项分布确定的靶的ml信号和sl误差的分离。
总之,这些数据验证了使用两个负二项分布的ml计数校正。
实例9
来自96孔混合jurkat和乳腺癌(brca)单细胞的bdprecisetm靶向测定的t-随机邻域嵌入可视化
该实例证明了基于对于混合jurkat和乳腺癌(brca)单细胞的递归取代错误校正和基于分布的错误校正来校正pcr和测序错误的方法。
图23a至23d示出了来自96孔混合的jurkat和乳腺癌(brca)单细胞(检查的86个基因)的precisetm靶向测定的示例性t-随机邻域嵌入(t-sne)可视化。图23a示出了在ml调整之前和之后,使用具有相同参数的dbscan来鉴定细胞簇。图23b至23d示出了通过颜色和点大小两者缩放的单个标志物表达。图23b示出了psmb4(管家基因),其存在于两种细胞类型中,并且在ml调整后,psmb4信号的缺乏在“低信号”簇中进一步突出显示。图23c示出了cd3e(突出jurkat细胞簇的淋巴细胞标志物)。图23d示出了cdh1(突出brca簇的上皮细胞标志物)。
总之,这些数据证明ml调节去除了ml噪音,其允许细胞簇之间的基因表达的明显区分。
实例10
细胞簇之间的差异表达分析
该实例证明了基于对于低信号细胞和乳腺癌(brca)细胞的递归取代错误校正和基于分布的错误校正来校正pcr和测序错误的方法。
图24a至24b是示出了在通过dbscan计算并通过每个簇中的基因标志物水平确定的两个选择的簇中具有>0个ml的基因的细胞簇之间的差异表达分析的非限制性示例性图。图24a示出了与剩余细胞相比的‘低信号’簇基因表达。图24a的顶部图示出了原始ml比较,这示出对于在其他细胞中具有较高平均表达的基因,ml噪音通常较高。图24a的底部图示出了在使用rsec和dbec进行ml调整之后,在“低信号”簇中检测到的ml噪音减少,这允许更清楚地区分簇之间的基因表达。图24b示出了与剩余细胞相比的‘brca’簇基因表达。图24b的顶部图示出了非brca细胞中的原始ml也具有brca标志物(诸如krt1、muc1)的显著的ml计数。图24b的底部图示出了brca标志物的经调整的ml在brca簇中比其他细胞中高度富集。
总之,这些数据证明了对于细胞(诸如低信号细胞和乳腺癌细胞),pcr和测序错误可以基于递归取代错误校正和基于分布的错误校正来校正。
实例11
调整混合的jurkat和t47d细胞的分子标记计数
此实例证明了调整混合的jurkat和t47d细胞的分子标记计数的方法。
图25a至25d是示出了来自96孔板的混合的jurkat和乳腺癌(t47d)单细胞(具有检查的86个基因)的bdprecisetm靶向测定的t-随机邻域嵌入(t-sne)可视化的非限制性示例性图。图25a示出了在ml调整之前和之后,使用具有相同参数的dbscan来鉴定细胞簇。图25b至25d示出了通过颜色和点大小缩放的单个标志物表达。图25b示出了psmb4的缩放(在两种细胞类型中和ml调整后存在的管家基因)。在无模板对照(ntc)簇中进一步突出了psmb4信号的缺乏。图25c示出了cd3e(突出jurkat细胞簇的淋巴细胞标志物)的缩放。图25d示出了cdh1(突出t47d簇的上皮细胞标志物)的缩放。
图26a至26b是在任何错误校正步骤(图26a中示出的原始ml)之前、以及在rsec和dbec校正(图26b中示出的经调整的ml)之后,通过图25a至25d中鉴定的不同细胞簇之间的分子标记计数显示差别基因表达的非限制性示例性热图。表达量低的基因是蓝色,且表达高的基因是橙色。在这些细胞类型之间基因表达模式相似的基因聚簇在一起。在没有校正错误的情况下,ntc具有来自高表达基因(如cd3e和krt18(其分别是jurkat和t47d标志物))的噪音。此外,错误校正揭示了jurkat与t47d之间不同的基因表达模式。
总之,这些数据证明ml调节可去除mi噪音,其允许细胞簇之间的基因表达的明显区分。
实例12
使用递归取代错误校正的免疫受体条形码错误校正
本实例证明了基于递归取代错误校正的免疫受体条形码错误校正。
图27a至27b示出了展示基于递归取代错误校正的免疫受体条形码错误校正的非限制性实例的表。进行免疫受体条形码错误校正包括通过递归取代错误校正来调整目的核苷酸序列(ns)(例如,cdr3的推定核苷酸序列)的计数。鉴定出cdr3的推定序列的多个簇。一个簇包括与亲本核苷酸序列
tgtgtggtgaacggagacggcactgccagtaaactcaccttt(seqidno:58)相差一个核苷酸(加下划线)的序列,诸如子核苷酸序列
tgtgtggtgaacggagacggcactgccagtaaactcactttt(seqidno:59)和
cgtgtggtgaacggagacggcactgccagtaaactcaccttt(seqidno:77)。核苷酸序列
tgtgtggtgaacggagacggcactgccagtaaactcaccttt(seqidno:58)是亲本核苷酸序列,因为它在此簇中具有最高原始读数为294,这是具有与测序数据中的此核苷酸序列相关联的不同序列的分子标记的数量。其他簇包括
tgtgctgtccaccgaggaagccaaggaaatctcatcttt(seqidno:78)和tgtgctgtccaccgaggaagccaaggaaatctcatcgtt(seqidno:79);tgtgcaggagaatctggggattaccagaaagttaccttt(seqidno:80)和
tgtgcaggagaatctgggggttaccagaaagttaccttt(seqidno:81);tgtgcagcaaccgagtcctatggtcagaattttgtcttt(seqidno:82)和tgtgcagcaacagagtcctatggtcagaattttgtcttt(seqidno:83)和
tgcctcgtggggagcctttctggttctgcaaggcaactgaccttt(seqidno:84)和
tgcctcgtggggagcctttccggttctgcaaggcaactgaccttt(seqidno:85)。每个子核苷酸序列的出现均归因于相对应的亲本分子标记序列(如从标记为“原始读数”的列到标记为“ns调整的读数”的列的箭头所示)。
进行免疫受体条形码错误校正包括通过递归取代错误校正来调整分子标记(ml)的计数。分子标记序列的簇包括亲本分子标记序列agtgcgag(seqidno:110)和子分子标记序列agtgcggg和agtgcnag(seqid号分别为111和112),它们与亲本分子标记序列相差一个核苷酸(加下划线)。在此实例中,与cdr3序列tgtgtggtgaacggagacggcactgccagtaaactcaccttt(seqidno:58)相关联的分子标记序列agtgcgag(seqidno:110)针对与分子标记agtgcgag(seqidno:110)相关联的所有cdr3序列具有最高核苷酸序列调整读数。每个子核苷酸序列的出现(子分子标记序列agtgcggg和agtgcnag(分别为seqidno:111和112)分别为两次和一次)归因于具有最高核苷酸序列调整读数(对于亲本分子标记agtgcgag(seqidno:110)为319)的相对应的亲本分子标记序列。进行免疫受体条形码错误校正包括基于rsec同时调整核苷酸序列和分子标记的计数。在此实例中,当应用rsec时,当每个核苷酸序列和相对应的分子标记被视为一个序列时,没有调整分子标记计数。
鉴定出多个嵌合体,并且去除了与嵌合体相对应的分子标记计数。例如,分子标记序列agtgcgag(seqidno:110)与多个cdr3核苷酸序列(诸如
tgtgtggtgaacggagacggcactgccagtaaactcaccttt(seqidno:58)和
tgtgctgtccaccgaggaagccaaggaaatctcatcttt(seqidno:78))相关联。核苷酸序列
tgtgctgtccaccgaggaagccaaggaaatctcatcttt(seqidno:78)的经调整的出现(例如,在上述调整后在测序数据中观察到的出现的数量)为7,低于核苷酸序列
tgtgtggtgaacggagacggcactgccagtaaactcaccttt(seqidno:58)的经调整的出现(为322),其是具有分子标记序列agtgcgag(seqidno:110)的所有核苷酸序列中最高的。去除与鉴定出的cdr3的嵌合序列相对应的靶的推定序列(如从标记为“ns和ml调整的读数”的列到标记为“去除的嵌合体”的列的箭头所示)。通过调整和去除嵌合体,将cdr3序列确定为
tgtgtggtgaacggagacggcactgccagtaaactcaccttt(seqidno.58),其与具有调整的分子标记计数为322的分子标记序列agtgcgag(seqidno.110)相关联。
总之,这些数据证明了用rsec调整核苷酸序列计数、分子标记计数以及核苷酸序列和分子计数,并且去除嵌合cdr3序列。
实例13
使用递归取代错误校正和基于分布的错误校正进行免疫受体条形码错误校正
本实例证明了使用递归取代错误校正和基于分布的错误校正进行免疫受体条形码错误校正。
将具有75%的健康外周血单核细胞(pbmc)和25%的jurkat细胞的500个细胞样品装载到rhapsodytm盒中,并且在包含rhapsodytm珠的微孔中捕获。rhapsodytm珠是磁珠,条形码附接在珠表面。每个珠都附有条形码,所述条形码具有细胞标记(其具有相同细胞标记序列)和分子标记(其选自一组不同分子序列)。不同的珠的条形码具有细胞标记,所述细胞标记具有不同细胞标记序列。每个珠的条形码具有用于捕获tcrmrna分子的捕获位点。如本文所描述的,对捕获的tcrmrna分子进行条形码化和测序。细胞标记和分子标记用于确定每个tcr分子的细胞和分子来源。使用分子标记确定不同tcr分子的出现的数量或计数。
使用免疫受体条形码错误校正(参照图7、图8、图27和实例12描述)校正不同tcr分子的测序数据。简而言之,基于递归取代错误校正来调整不同的tcr基因(例如,tcrb)的推定核苷酸序列的计数。基于递归取代错误校正来调整分子标记的计数。基于递归取代错误校正来调整核苷酸序列(例如,tcrb)和分子标记的计数。去除与嵌合体相对应的分子标记计数。随后,使用本文所描述的基于分布的错误校正来调整分子标记计数。图28是示出了对于tcrb进行免疫受体条形码错误校正然后进行基于分布的错误校正的非限制性示例性结果的直方图。如果不进行错误校正,tcr多样性(包括tcrb多样性)将被过高估计。
总之,这些数据证明了用递归取代错误校正调整tcr核苷酸序列计数、分子标记计数以及tcr核苷酸序列和分子计数,并且去除嵌合tcr序列,然后进行基于分布的错误校正以避免对tcr多样性的过高估计。
在至少一些先前描述的实施例中,在一个实施例中使用的一个或多个元素可以互换地用于另一个实施例中,除非这种替换在技术上不可行。本领域技术人员将理解,在不脱离所要求保护的主题的范围的情况下,可以对上述方法和结构进行各种其他的省略、添加和修改。所有此类修改和改变都旨在落在由所附权利要求书限定的主题的范围内。
关于本文中使用基本上任何复数和/或单数术语,在对于背景和/或应用适当的情况下,本领域技术人员可以从复数转换为单数和/或从单数转换为复数。为了清楚起见,可以在本文明确阐述各种单数/复数排列。如本说明书和所附权利要求书中使用的,除非上下文另有明确指示,否则单数形式“一个/一种(a/an)”和“所述(the)”包括复数的提及物。除非另外说明,在本文中对“或”的任何提及旨在涵盖“和/或”。
本领域技术人员将理解,一般来说,本文使用的术语,尤其是所附权利要求书(例如,所附权利要求书的主体)中的术语,通常旨在作为“开放性的”术语(例如,术语“包括(including)”应解释为“包括但不限于(includingbutnotlimitedto)”,术语“具有(having)”应解释为“具有至少(havingatleast)”,术语“包括(includes)”应解释为“包括但不限于(includesbutisnotlimitedto)”等)。本领域技术人员将进一步理解,如果预期到所介绍的权利要求陈述的特定数目,这样的预期将明确地陈述于权利要求中,并且在不存在这种陈述的情况下没有这种意图存在。例如,作为对理解的帮助,以下所附权利要求书可以包含介绍性短语“至少一个”和“一个或多个”的使用,以介绍权利要求陈述。然而,此类短语的使用不应解读为意味着由不定冠词“一个”或“一种”介绍权利要求陈述会将任何包含这种介绍的权利要求陈述的具体权利要求限制到包含仅一个这种陈述的实施例中,甚至当相同的权利要求包括介绍性短语“一个或多个”或“至少一个”以及不定冠词如“一个”或“一种”时也是如此(例如,“一个”和/或“一种”应解释为意指“至少一个”或“一个或多个”);这对于使用定冠词来介绍权利要求陈述同样适用。此外,即使明确地陈述了介绍的权利要求陈述的特定数目,本领域技术人员将认识到,这种陈述应解释为意指至少所陈述的数字(例如,仅陈述“两个陈述”而没有其他修饰词意指至少两个陈述、或两个或更多个陈述)。此外,在使用类似于“a、b和c等中的至少一个”的惯例的那些情况下,通常这种句法结构是在本领域技术人员将理解该惯例的意义上预期(例如,“具有a、b和c中的至少一个的系统”将包括但不限于仅具有a,仅具有b,仅具有c,a和b一起,a和c一起,b和c一起,和/或a、b、和c一起等的系统)。在使用类似于“a、b或c等中的至少一个”的惯例的那些情况下,通常这种句法结构是在本领域技术人员将理解该惯例的意义上预期(例如,“具有a、b或c中的至少一个的系统”将包括但不限于仅具有a,仅具有b,仅具有c,a和b一起,a和c一起,b和c一起,和/或a、b、和c一起等的系统)。本领域技术人员将进一步理解,实际上,无论在说明书、权利要求书还是在附图中,呈现两个或更多个替代术语的任何分离性词语和/或短语应被理解为考虑到包括术语之一、任一术语或两个术语的可能性。例如,短语“a或b”将被理解为包括“a”或“b”或“a和b”的可能性。
此外,当本披露的特征或方面以马库什组(markushgroup)描述时,本领域技术人员将意识到本披露还由此以马库什组的任何单独的成员或成员子组描述。
如本领域技术人员将理解的,出于任何和所有目的,如在提供书面描述方面,本文披露的所有范围还包括任何和所有可能的它的子范围和子范围组合。任何列出的范围都可以很容易地被识别为充分描述并使相同的范围能被分解为至少相等的一半,三分之一,四分之一,五分之一,十分之一等。作为非限制性实例,这里讨论的每个范围可以容易地分解为下三分之一,中三分之一和上三分之一等。如本领域技术人员还将理解的,所有语言,如“多至”、“至少”、“大于”、“小于”等包括所陈述的数字,并且指代可以随后分解为如上讨论的子范围的范围。最后,如本领域技术人员将理解的,范围包括每个单独的成员。因此,例如,具有1-3个物品的组是指具有1、2或3个物品的组。类似地,具有1-5个物品的组指代具有1、2、3、4或5个物品的组,等等。
尽管本文已经披露了各种方面和实施例,但其他方面和实施例对本领域技术人员将是明显的。本文披露的各种方面和实施例用于说明的目的而并不意于限制由以下权利要求所指出的真实范围和精神。
序列表
<110>赛卢拉研究公司
艾琳·夏姆
范珏
詹尼弗·蔡
<120>免疫受体条形码错误校正
<130>bdcri.035wo
<150>62/562,978
<151>2017-09-25
<160>113
<170>patentin3.5版
<210>1
<211>20
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>1
aaaaaaaaaaaaaaaaaaaa20
<210>2
<211>20
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>2
tttttttttttttttttttt20
<210>3
<211>8
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>3
gtcaaatt8
<210>4
<211>8
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>4
gtcaaaat8
<210>5
<211>8
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>5
gtcaaaaa8
<210>6
<211>8
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>6
ttcaaaaa8
<210>7
<211>8
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>7
ttcagaaa8
<210>8
<211>8
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>8
ctcaaaaa8
<210>9
<211>8
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>9
ttcaaact8
<210>10
<211>8
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>10
ttcaaaat8
<210>11
<211>8
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>11
ttcaaaca8
<210>12
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>12
cysvalvalasnglyaspglythralaserlysleuthrphe
1510
<210>13
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>13
cysvalvalaspglyaspglythralaserlysleuthrphe
1510
<210>14
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>14
cysvalvalserglyaspglythralaserlysleuthrphe
1510
<210>15
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>15
cysvalvalasnglyvalglythralaserlysleuthrphe
1510
<210>16
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>16
cysvalvalasnglyaspglyalaalaserlysleuthrphe
1510
<210>17
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>17
cysvalvalasnglyaspglythralaglylysleuthrphe
1510
<210>18
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>18
cysvalvalasnglyaspglythralaserargleuthrphe
1510
<210>19
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>19
cysvalvalasnglyaspglythralaserlysleualaphe
1510
<210>20
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>20
cysvalvalasnglyaspglythralaserlysleuthrleu
1510
<210>21
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>21
cysvalvalasnglyaspglythralaserlysprothrphe
1510
<210>22
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>22
cysvalvalasnglyaspglythrthrserlysleuthrphe
1510
<210>23
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>23
cysvalvalasngluaspglythralaserlysleuthrphe
1510
<210>24
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>24
cysvalalaasnglyaspglythralaserlysleuthrphe
1510
<210>25
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>25
cysvalmetasnglyaspglythralaserlysleuthrphe
1510
<210>26
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>26
cysalavalasnglyaspglythralaserlysleuthrphe
1510
<210>27
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>27
argvalvalasnglyaspglythralaserlysleuthrphe
1510
<210>28
<211>13
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>28
cysalavalhisargglyserglnglyasnleuilephe
1510
<210>29
<211>13
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>29
cysalavalhisargglyserglnglyasnleuileval
1510
<210>30
<211>13
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>30
cysalaglygluserglyasptyrglnlysvalthrphe
1510
<210>31
<211>13
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>31
cysalaglygluserglyglytyrglnlysvalthrphe
1510
<210>32
<211>13
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>32
cysalaalathrglusertyrglyglnasnphevalphe
1510
<210>33
<211>15
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>33
cysleuvalglyserleuserglyseralaargglnleuthrphe
151015
<210>34
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>34
cysvalvalthralaserglyglytyrglnlysvalthrphe
1510
<210>35
<211>13
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>35
cysalavalalaprotyrglyasnasnargleualaphe
1510
<210>36
<211>15
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>36
cysalavalthrargpheserglyglytyrasnlysleuilephe
151015
<210>37
<211>17
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>37
cysalavalserlysglyalaargserglyasnthrglylysleuile
151015
phe
<210>38
<211>10
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>38
cysalaleupheasnasnaspmetargphe
1510
<210>39
<211>13
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>39
cysalaleuserproglyglytyrglnlysvalthrphe
1510
<210>40
<211>15
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>40
cysalaglyleulysleugluthrserglyserargleuthrphe
151015
<210>41
<211>11
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>41
cysalaglyglytyrglyasnlysleuvalphe
1510
<210>42
<211>16
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>42
cysalaglyalaargglyserasnpheglyasnglulysleuthrphe
151015
<210>43
<211>12
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>43
cysalaalaasnasnalaglyasnvalleuthrphe
1510
<210>44
<211>15
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>44
cysalaalaproserleuglyglyseralaargglnleuthrphe
151015
<210>45
<211>15
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>45
cysalaalaserileargglyaspsersertyrlysleuilephe
151015
<210>46
<211>13
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<220>
<221>对应于终止密码子的位置
<222>(7)..(8)
<400>46
cysalaalaserargalaaspglyasnglnphetyrphe
1510
<210>47
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>47
cysalaalaserprometasnargaspasplysileilephe
1510
<210>48
<211>14
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>48
cysalaalaserilethraspsertrpglylysleuglnphe
1510
<210>49
<211>13
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>49
cysvalvalseralalysasnthrasplysleuilephe
1510
<210>50
<211>12
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>50
cysalatyrargserserasntyrglnleuiletrp
1510
<210>51
<211>15
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>51
cysalavalvalpropheglyglyglyglyasnlysleuthrphe
151015
<210>52
<211>12
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>52
cysalaglytrpserasnasptyrlysleuserphe
1510
<210>53
<211>13
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>53
cysalaalaserglyglyserasntyrlysleuthrphe
1510
<210>54
<211>16
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>54
cysalametarggluglyglyglyserasnasptyrlysleuserphe
151015
<210>55
<211>13
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>55
cysileargleuproglyasnthrglylysleuilephe
1510
<210>56
<211>13
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>56
cysalatyrvalalaalaalaglyasnlysleuthrphe
1510
<210>57
<211>12
<212>prt
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>57
cysalaglyalaproglysertyrileprothrphe
1510
<210>58
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>58
tgtgtggtgaacggagacggcactgccagtaaactcaccttt42
<210>59
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>59
tgtgtggtggacggagacggcactgctagtaaactcaccttt42
<210>60
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>60
tgtgtggtgagcggagacggcactgccagtaaactcaccttt42
<210>61
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>61
tgtgtggtgaacggagtcggcactgccagtaaactcaccttt42
<210>62
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>62
tgtgtggtgaacggagacggcgctgccagtaaactcaccttt42
<210>63
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>63
tgtgtggtgaacggagacggcactgccggtaaactcaccttt42
<210>64
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>64
tgtgtggtgaacggagacggcactgccagtagactcaccttt42
<210>65
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>65
tgtgtggtgaacggagacggcactgccagtaaactcgccttt42
<210>66
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>66
tgtgtggtgaacggagacggcactgccagtaaactcactttt42
<210>67
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>67
tgtgtggtgaacggagacggcactgccagtaaactcaccctt42
<210>68
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>68
tgtgtggtgaacggagacggcactgccagtaaacccaccttt42
<210>69
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>69
tgtgtggtgaacggagacggcactgccagcaaactcaccttt42
<210>70
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>70
tgtgtggtgaacggagacggcactaccagtaaactcaccttt42
<210>71
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>71
tgtgtggtgaacggagacggcacagccagtaaactcaccttt42
<210>72
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>72
tgtgtggtgaacgaagacggcactgccagtaaactcaccttt42
<210>73
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>73
tgtgtggcgaacggagacggcactgccagtaaactcaccttt42
<210>74
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>74
tgtgtgatgaacggagacggcactgccagtaaactcaccttt42
<210>75
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>75
tgtgcggtgaacggagacggcactgccagtaaactcaccttt42
<210>76
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>76
tgcgtggtgaacggagacggcactgccagtaaactcaccttt42
<210>77
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>77
cgtgtggtgaacggagacggcactgccagtaaactcaccttt42
<210>78
<211>39
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>78
tgtgctgtccaccgaggaagccaaggaaatctcatcttt39
<210>79
<211>39
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>79
tgtgctgtccaccgaggaagccaaggaaatctcatcgtt39
<210>80
<211>39
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>80
tgtgcaggagaatctggggattaccagaaagttaccttt39
<210>81
<211>39
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>81
tgtgcaggagaatctgggggttaccagaaagttaccttt39
<210>82
<211>39
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>82
tgtgcagcaaccgagtcctatggtcagaattttgtcttt39
<210>83
<211>39
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>83
tgtgcagcaacagagtcctatggtcagaattttgtcttt39
<210>84
<211>45
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>84
tgcctcgtggggagcctttctggttctgcaaggcaactgaccttt45
<210>85
<211>45
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>85
tgcctcgtggggagcctttccggttctgcaaggcaactgaccttt45
<210>86
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>86
tgtgtggtgaccgcttctgggggttaccagaaagttaccttt42
<210>87
<211>39
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>87
tgtgctgtggccccctatgggaacaacagactcgctttt39
<210>88
<211>45
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>88
tgtgctgtgactcggttttctggtggctacaataagctgattttt45
<210>89
<211>51
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>89
tgtgctgtcagtaagggggctaggtctggcaacacaggcaaactaatcttt51
<210>90
<211>30
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>90
tgtgctctgtttaacaatgacatgcgcttt30
<210>91
<211>39
<212>dna
<213>人工序列
<220>
<223>注释=“工序列的描述:合成
寡核苷酸或多肽”
<400>91
tgtgctctgtcccctgggggttaccagaaagttaccttt39
<210>92
<211>45
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>92
tgtgcagggttaaaactagaaaccagtggctctaggttgaccttt45
<210>93
<211>33
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>93
tgtgcaggggggtatggaaacaaactggtcttt33
<210>94
<211>48
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>94
tgtgcaggagcgaggggatctaactttggaaatgagaaattaaccttt48
<210>95
<211>36
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>95
tgtgcagctaataatgcaggcaacgtgctcaccttt36
<210>96
<211>45
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>96
tgtgcagccccctccctggggggttctgcaaggcaactgaccttt45
<210>97
<211>45
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>97
tgtgcagcaagtataaggggggatagcagctataaattgatcttc45
<210>98
<211>40
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>98
tgtgcagcaagtagagccgaccggtaaccagttctatttt40
<210>99
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>99
tgtgcagcaagcccaatgaacagagatgacaagatcatcttt42
<210>100
<211>42
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>100
tgtgcagcaagcataactgacagctgggggaaattgcagttt42
<210>101
<211>39
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>101
tgtgtggtgagcgcgaaaaacaccgacaagctcatcttt39
<210>102
<211>36
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>102
tgtgcttataggagtagcaactatcagttaatctgg36
<210>103
<211>45
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>103
tgtgctgtggtccccttcgggggaggaggaaacaaactcaccttt45
<210>104
<211>36
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>104
tgtgcaggatggtctaacgactacaagctcagcttt36
<210>105
<211>39
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>105
tgtgcagcaagtggaggtagcaactataaactgacattt39
<210>106
<211>48
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>106
tgtgcaatgagagagggcggtggttctaacgactacaagctcagcttt48
<210>107
<211>39
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>107
tgcatccgcctgcctggcaacacaggcaaactaatcttt39
<210>108
<211>39
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>108
tgtgcttatgtcgcagctgcaggcaacaagctaactttt39
<210>109
<211>36
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>109
tgtgcaggagccccaggaagctacatacctacattt36
<210>110
<211>8
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>110
agtgcgag8
<210>111
<211>8
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>111
agtgcggg8
<210>112
<211>8
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<220>
<221>混杂特征
<222>(6)..(6)
<223>n是a,c,g,或t
<400>112
agtgcnag8
<210>113
<211>8
<212>dna
<213>人工序列
<220>
<223>注释=“人工序列的描述:合成
寡核苷酸或多肽”
<400>113
atagagat8
- 还没有人留言评论。精彩留言会获得点赞!