转录组解析装置及解析方法与流程
本发明涉及对关于转录组的信息进行解析的转录组解析装置及解析方法。
背景技术:
作为基于基因表达而预测生物的表现型的尝试,已知从基因表达数据和表现型数据重回归分析的方法(非专利文献1及专利文献1)。在非专利文献1中公开的方法中,为了使基因表达数据的重复消失,对于相同的操纵子而应用仅表达水平最高的数据等而限定基因表达数据。
即便如此,转录组,一般而言,是指存在于指定的状态或条件下的组织或细胞内的全部转录产物。转录组含来自基因组上的编码区域的转录产物(即,mrna)和来自非编码区域的转录产物(所谓的ncrna)。通过对转录组进行解析,可得到因环境要因导致的基因表达的变动、与表现型关联表达的基因的鉴定等基于基因的表达状态的新的见解。
在解析转录组时,例如,将存在于组织或细胞内的转录产物应用微阵列技术或下一代测序技术而整体测量。测量的数据是大量的碱基序列数据,是典型的大数据。
作为统计学解析得到的数据的方法,如在专利文献2中公开,已知应用作为多变量解析的一方法的主成分分析的方法。在所述方法中,通过对于训练数据(不是由分析得到的碱基序列数据)进行主成分分析,可导出在条件不同的样品间能比较的结果。
另外,作为转录组解析法,如在专利文献3中公开,已知从基因表达信息(状态变量)和性状信息(特性变量)生成解析对象的特性变量推定模型的方法。在专利文献3中公开的方法中,以特性变量作为目的变量(从属变量)、以状态变量的各自作为说明变量,生成有正则化项的回归模型。作为回归模型的算出式,例示lasso回归(leastabsoluteshrinkageandselectionoperator)。
即便如此,lasso回归是指为了防在统计学或机器学习的领域中的过度拟合而使用的正则化的一方法(l1型正则化法),是将大量的数据之中不重要的数据的参数设为0,从数据删除的基于稀疏正则化法的回归建模(非专利文献2)。
【现有技术文献】
【专利文献】
【专利文献1】wo2016/148107
【专利文献2】专利第5854346号
【专利文献3】特开2017-51118号公报
【非专利文献】
【非专利文献1】naturecommunications5,articlenumber:5792(2014)
【非专利文献2】roberttibshirani,journaloftheroyalstatisticalsociety.seriesb(methodological)vol.58,no.1(1996),pp.267-288
【发明的概要】
【发明要解决的课题】
即便如此,由于在上述的转录组解析中,与解析对象的样品数比较,得到了碱基序列数据的转录产物的数极其大,因此在非专利文献1中公开的方法中难以得到充分地有含意的解析结果。另外,对于应用在专利文献3中公开的lasso回归分析的解析方法,即使在与解析对象的样品数比较而得到碱基序列数据的转录产物的数是极其大时也期待良好的解析结果。但是,在转录组解析中,要求解析结果的进一步的精度提升。
从而,本发明鉴于上述的实情,旨在提供可使用关于转录产物的碱基序列数据而进行更高精度的转录组解析的转录组解析装置及解析方法。
【用于解决课题的手段】
达成上述的目的的本发明包含以下。
(1)转录组解析装置,其具备:生成对于含目的变量数据和基因表达量数据的多个数据集随机地削减基因表达量数据的第1~第m的子数据集(m≥2)的数据集生成单元,生成对于第1~第m的子数据集各自应用有正则化项的回归分析法而算出以目的变量数据作为目的变量、以基因表达量数据作为说明变量的第1~第m的预测式的预测式算出单元,及生成对应于第1~第m的预测式中所含的基因表达量数据的基因的列表的基因列表生成单元。
(2)(1)所述的转录组解析装置,其特征在于,上述预测式算出单元作为上述回归分析法而应用lasso(leastabsoluteshrinkageandselectionoperator)。
(3)(1)所述的转录组解析装置,其特征在于,上述数据集生成单元生成1000~20000轮的子数据集(m=1000~20000)。
(4)(1)所述的转录组解析装置,其特征在于,上述基因列表生成单元基于第1~第m的预测式而算出基因的出现概率,与算出的出现概率相关联而生成基因的列表。
(5)(1)所述的转录组解析装置,其特征在于,上述基因列表生成单元从储存了基因的注释信息的数据库读取列表中所含的基因的注释信息,与读取的注释信息相关联而生成基因的列表。
(6)(1)所述的转录组解析装置,其特征在于还有对于由上述基因列表生成单元生成的列表中所含的多个基因,由使用上述多个数据集中所含的目的变量数据和基因表达量数据的重回归分析生成关于指定的目的变量的预测模型式的预测模型式生成单元。
(7)转录组解析方法,其包括:生成对于含目的变量数据和基因表达量数据的多个数据集随机地削减基因表达量数据的子数据集的子数据集生成工序,对于子数据集应用正则化法而算出以目的变量数据作为目的变量、以基因表达量数据作为说明变量的预测式的预测式算出工序,记录对应于预测式中所含的基因表达量数据的基因的基因记录工序,及将上述子数据集生成工序、上述预测式算出工序及上述基因记录工序重复m次(m≥2),生成记录的基因的列表的基因列表生成工序。
(8)(7)所述的转录组解析方法,其特征在于,在上述预测式算出工序中,作为上述正则化法而应用lasso(leastabsoluteshrinkageandselectionoperator)。
(9)(7)所述的转录组解析方法,其特征在于,在上述子数据集生成工序中,生成1000~20000轮的子数据集(n=1000~20000)。
(10)(7)所述的转录组解析方法,其特征在于,在上述基因列表生成工序中,基于以第1~第m次的重复生成的第1~第m的预测式而算出基因的出现概率,与算出的出现概率相关联而生成基因的列表。
(11)(7)所述的转录组解析方法,其特征在于,在上述基因列表生成工序中,从储存了基因的注释信息的数据库读取列表中所含的基因的注释信息,与读取的注释信息相关联而生成基因的列表。
(12)(7)所述的转录组解析方法,其特征在于,在上述基因列表生成工序之后还有,对于生成的列表中所含的多个基因,由使用上述多个数据集中所含的目的变量数据和基因表达量数据的重回归分析生成关于指定的目的变量的预测模型式的预测模型式生成工序。
【发明的效果】
根据本发明涉及的转录组解析装置及解析方法,能进行关于转录组的高精度的解析。从而,通过应用本发明涉及的转录组解析装置及解析方法,可高精度地进行例如,由指定的状态或条件这样的要因导致的基因表达的变动解析、与表现型关联的基因的表达解析、或者,基于基因表达的性状的预测解析等。
【附图说明】
【图1】是显示本发明涉及的转录组解析装置的一实施方式的功能框图。
【图2】是显示本发明涉及的转录组解析方法的一实施方式的流程图。
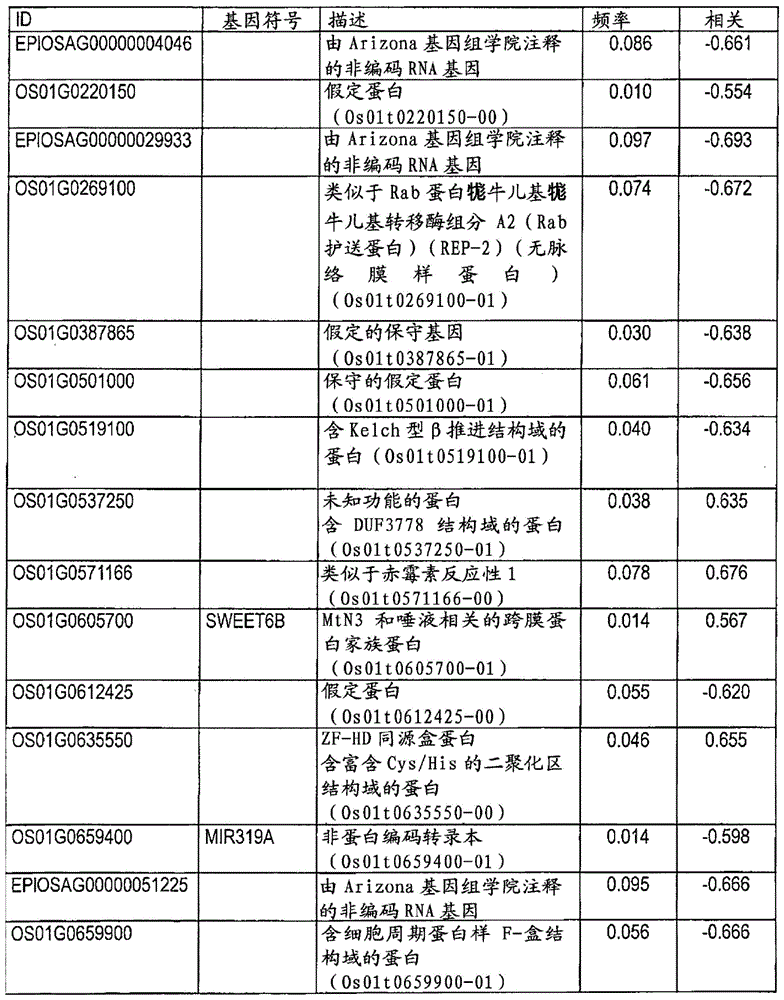
【图3】是显示由转录组解析装置及解析方法输出的基因的列表的一例的特性图。
【图4】是显示由转录组解析装置及解析方法输出的基因的列表的其他例的特性图。
【图5】是显示本发明涉及的转录组解析装置的其他实施方式的功能框图。
【图6】是显示本发明涉及的转录组解析方法的其他实施方式的流程图。
【图7】是显示由转录组解析装置及解析方法输出的预测值和实测值的关系的特性图。
【图8】是显示应用本发明的预测评价系统的构成的框图。
【图9】是对arrozdaterra和ouu365的发芽14天后进行摄像的照片。
【图10】是显示使用bil104系统而进行地上部干物重的qtl解析的结果的特性图。
【图11】是显示实施例中制出的系统的数和地上部干燥重量的关系的特性图。
【图12】是对于实施例中制出的系统而显示地上部生鲜重的特性图。
【图13-1】是显示在实施例中作为表达量生物标志物以高频度选出的158个基因的列表的特性图。
【图13-2】是显示在实施例中作为表达量生物标志物以高频度选出的158个基因的列表的特性图。
【图13-3】是显示在实施例中作为表达量生物标志物以高频度选出的158个基因的列表的特性图。
【图13-4】是显示在实施例中作为表达量生物标志物以高频度选出的158个基因的列表的特性图。
【图13-5】是显示在实施例中作为表达量生物标志物以高频度选出的158个基因的列表的特性图。
【图13-6】是显示在实施例中作为表达量生物标志物以高频度选出的158个基因的列表的特性图。
【图13-7】是显示在实施例中作为表达量生物标志物以高频度选出的158个基因的列表的特性图。
【图13-8】是显示在实施例中作为表达量生物标志物以高频度选出的158个基因的列表的特性图。
【图13-9】是显示在实施例中作为表达量生物标志物以高频度选出的158个基因的列表的特性图。
【图14】是对于在实施例中制出的rna-seq解析中使用的bil系统和亲本品种而显示qltg3-1表达量和地上部生鲜重的关系的特性图。
【图15】是对于在实施例中制出的rna-seq解析中使用的bil系统和亲本品种而显示sg-1表达量和地上部生鲜重的关系的特性图。
【图16】是对于实施例中制出的bil104系统全部和亲本品种而显示sg-1表达量和地上部生鲜重的关系的特性图。
【图17】是显示从关于在实施例1中制成的列表中所含的基因的表达量数据及地上部生鲜重数据算出的地上部生鲜重的预测值和地上部生鲜重的实测值的关系的特性图。
【具体实施方式】
以下,对本发明涉及的转录组解析装置及/或解析方法参照附图而详细地进行说明。
〔第1实施方式〕
本发明涉及的转录组解析装置1如图1所示,具备:对于指定的目的变量数据而从含多数的基因表达量数据(p维、其中p相当于转录产物的数)的数据集生成第1~第m的数据集(2≤m≤p-1)的数据集生成部2,对于在数据集生成部2中生成的第1~第m的数据集各自应用正则化法而算出以目的变量数据作为目的变量、以基因表达量数据作为说明变量的第1~第m的预测式的预测式算出部3,及生成对应于在预测式算出部3算出的第1~第m的预测式中所含的基因表达量数据的基因的列表的基因列表生成部4。另外,转录组解析装置1也可为可接入到储存了基因的注释信息的外部的数据库5的装置。
输入到转录组解析装置1的数据集含指定的目的变量数据和基因表达量数据(p维)。其中,目的变量数据是指含关于包括量的性状或质的性状的表现型的数值数据、关于周边环境条件的有无以及程度的数值数据、关于对于解析对象生物的处理的有无以及程度的数值数据的含意。更具体性地,目的变量数据可举关于作为植物体的解析对象生物的生长量的数据(例如,地上部重量、根部重量、叶面积、种子收量等)、关于负载在解析对象生物的应激的数据(例如,高温度处理时间、低温度处理时间、药剂处理浓度、病害虫应激时间等)。
另外,基因表达量数据是指对于观察到的转录产物而显示表达量的相对量的数值数据。更具体性地,作为基因表达量数据,可举市售的基因表达解析用(转录组解析用)微阵列,或利用市场提供的基因表达解析保藏服务等而得到的微阵列数据或,利用使用下一代测序装置的表达解析(rna-seq)而得到的序列数据等。特别是,作为基因表达量数据,优选采用利用使用下一代测序装置的表达解析(rna-seq)而得到的序列数据。这是因为,在利用使用下一代测序装置的表达解析(rna-seq)而得到的序列数据中,包括在解析对象生物中的转录产物。
采用转录组解析装置1,通过对上述数据集进行解析,可生成可对目的变量数据进行说明的基因的列表。再者,基因的列表是指不限定于编码蛋白质的狭义的基因的列表,也含来自非编码区域的转录产物的列表的含意。
例如,在以植物体的初期生长量(指定的期间的植物重量)作为目的变量数据时,由转录组解析装置1可生成可对初期生长量进行说明的基因列表。另外,在以向植物体处理的药剂浓度作为目的变量数据时,由转录组解析装置1可生成与处理的药剂浓度关联而表达的基因的列表。再者,在以采样时的气温作为目的变量数据时,由转录组解析装置1可生成与生长温度关联而表达的基因的列表。
图1中所示的构成的转录组解析装置1可根据例如图2中所示的流程图而生成上述基因的列表。
首先,输入从微阵列装置或下一代测序装置等输出的基因表达量数据和目的变量数据(步骤s1)。其中,以输入的基因表达量数据作为p维,作为p维说明变量向量x={x1,……,xp}。另外,以输入的目的变量作为y。再者,在本例中,输入由p维说明变量向量x和目的变量y组成的n组的数据集({(yi,xi)|i=1,……,n})。
接下来,在数据集生成部2中,通过随机地采样输入的n组的数据集中所含的基因表达量数据,生成p-1维以下的子数据集(步骤s2)。再者,在本步骤中,生成将初始值设为m=1的第m的子数据集。换言之,生成的第m的子数据集定义为随机地削减输入的数据集中所含的基因表达量数据而含比输入的数据集少的数的基因表达量数据的数据集。
其中,生成的第m的子数据集只要是输入的数据集中所含的基因表达量数据(p维)的一部分即可,例如,可设为p维的基因表达量数据之中5~90%的数据,可设为5~70%的数据,可设为5~50%的数据,可设为10~50%的数据,可设为10~25%的数据,可设为10~15%的数据。
例如,在基因表达量数据数是30000时(即p=30000、30000个转录产物),在数据集生成部2中生成的第m的子数据集可含随机地选择的1000~20000个基因表达量数据,优选为可含1500~15000个基因表达量数据,更优选为可含1500~7500个基因表达量数据,再优选为可含1500~4500个基因表达量数据。
接下来,在预测式算出部3中,对于在数据集生成部2中生成的第m的子数据集应用有正则化项的回归分析法而算出以目的变量数据作为目的变量、以基因表达量数据作为说明变量的第m的预测式(步骤s3)。其中,有正则化项的回归分析法也称为正则化回归模型,是指向最小二乘法付加制约(罚则)而使推定量缩小的解析法。具体而言,作为有正则化项的回归分析法,可举lasso回归分析法、ridge回归分析法及弹性网回归分析法。特别是,优选应用lasso回归分析法而算出预测式。在本步骤中算出的预测式特别在应用lasso回归分析法时,成为将说明目的变量之时不重要的基因表达量数据的参数设为0的预测式。
再者,在应用lasso回归分析法而算出预测式时,可参照friedmanetal.,regularizationpathsforgeneralizedlinearmodelsviacoordinatedescent,journalofstatisticalsoftware,january2010,volume33,issue1。
接下来,在基因列表生成部4中,提取在预测式算出部3中算出的预测式所含的基因表达量数据,记录对应于提取的基因表达量数据的基因(步骤s4)。即,由于预测式由有正则化项的回归分析法算出,因此在说明目的变量之时,可仅提取重要的基因表达量数据。例如,在作为有正则化项的回归分析法而应用lasso回归分析法时,提取将参数设为0的基因表达量数据以外的基因表达量数据。
接下来,在步骤s5中,判断是否将上述步骤s2~s4的处理重复预先规定的次数(m次)。例如,在作为重复次数而预先规定10000次(m=10000)时,对于将初始值设为1的第1子数据集执行上述步骤s2~s4的处理之后,在步骤s5中将m=1的值与10000进行比较,进到步骤s6。在步骤s6中使m值增加1,重复步骤s2~s5至m值成为10000。
通过将以上的步骤s2~s6重复m次,可对于在步骤s1中输入的n组的数据集而对于第1~m的子数据集各自而算出第1~第m的预测式。
接下来,在步骤s7中,在基因列表生成部4中,对于第1~m的预测式而作为列表输出在步骤s4中记录的基因。在基因列表生成部4中生成的基因的列表不特别限定,可为列举对应于用基因列表生成部4提取的基因表达量数据的基因的形式,也可为将对应于提取的基因表达量数据的基因和所述基因的出现概率相关联的形式。其中出现概率可作为相对于第1~第m的预测式中所含的全部基因数而含特定的基因的次数算出。
另外,在基因列表生成部4中生成的基因的列表可为仅含如上所述算出的出现概率超指定的值的基因的形式,也可为从如上所述算出的出现概率高的基因顺序地列举的形式。
作为一例,在基因列表生成部4中生成的基因的列表的输出例示于图3。在基因列表生成部4中生成的基因的列表如图3所示,含针对每个转录产物分配的id、关于转录产物所来源的基因的符号、针对每个转录产物算出的出现概率、及与目的变量数据的相关系数。
再者,也可如图1所示,转录组解析装置1接入外部的数据库5,检索列表中所含的基因的注释信息等,以得到的注释信息等作为与基因关联的形式。另外,转录组解析装置1也可基于检索的注释信息而对基因进行分组,以每组基因作为列表的形式。
作为一例,在基因列表生成部4中生成的基因的列表的输出例示于图4。在基因列表生成部4中生成的基因的列表如图4所示,含:针对每个转录产物分配的id、关于转录产物所来源的基因的符号、关于用所述基因符号确定的基因的功能的信息、针对每个转录产物算出的出现概率、及与目的变量数据的相关系数。
根据图3及/或4中所示的基因列表,作为关于指定的目的变量数据而解析的结果,可理解可对所述目的变量数据进行说明的基因组。特别是上述出现概率与这些基因列表关联时,对于列表中所举的各基因,可基于所述出现概率而判断与目的变量数据的关联性的强度。再者,在注释信息与这些基因列表相关联时,可对于列表中所举的各基因,基于所述注释信息而对于与目的变量数据的关联性理解生物学的含意。
〔第2实施方式〕
即便如此,本发明涉及的转录组解析装置及解析方法不限于上述的第1实施方式,也可为如图5及6所示,利用关于指定的目的变量数据制成的基因的列表而制成关于所述目的变量数据的预测模型式。再者,在图5及6中所示的转录组解析装置10及解析方法中,通过对于与图1及2中所示的转录组解析装置及解析方法相同的构成及工序赋予与图1及2相同的符号而省略对其详细的说明。
图5中所示的转录组解析装置10具备基于在基因列表生成部4中生成的列表中所含的基因而生成预测模型式的预测模型式生成部11。具备预测模型式生成部11的转录组解析装置可生成除了在基因列表生成部4中生成的基因的列表(例如图3及4)之外,还含对指定的目的变量数据进行说明的说明变量的预测模型式。
在转录组解析装置10中,如图6所示,与上述的第1实施方式同样地,由步骤s1~s6生成基因列表。其后,在转录组解析装置10中,在预测模型式生成部11中,对于列表中所含的基因,从在步骤s1中输入的n个数据集各自读取关于所述基因的说明变量数据及目的变量数据。进而,可由使用关于各基因的目的变量y及说明变量x的值的重回归分析或机器学习来构建说明指定的目的变量数据的预测模型式。
另外,在预测模型式生成部11中,可由使用关于在基因列表生成部4中生成的列表中所含的全部基因的目的变量y及说明变量x的值的重回归分析或机器学习来生成说明指定的目的变量数据的预测模型式,也可由使用关于所述列表中所含的一部分的基因的目的变量y及说明变量x的值的重回归分析或机器学习来生成说明指定的目的变量数据的预测模型式。作为列表中所含的一部分的基因,例如,可采用出现频度的值超阈值的范围的基因,也可采用指定的注释信息关联的基因。
作为用于构建预测式的方法,不特别限定,可举例如,选自lasso回归解析法、ridge回归解析法及弹性网解析法等的重回归法,或随机森林法及深度学习等的机器学习法。
作为一例,可在预测模型式生成部11中应用随机森林法而制成预测模型式。用此随机森林法制成的预测模型式是算出指定的目的变量y的决策树的形式的模型式,作为在基因列表生成部4中生成的列表中所含的基因的基因表达量数据x的函数生成。根据在预测模型式生成部11中生成的预测模型式,可基于对于指定的生物取得的基因表达量数据而关于所述生物算出目的变量的预测值。
其中,在基因列表生成部4中生成的列表中所含的目的变量y(实测值)和基于应用随机森林法而制成的预测模型式算出的预测值的关系示于图7。如图7所示得知,根据应用随机森林法制成的预测模型式,算出的预测值与实测值显示非常地高的拟合度。再者,图7中所示的坐标图使用记载在后述的实施例中的数据。
在以例如植物的种子收量作为目的变量而得到上述预测模型式时,通过使用从检查对象的植物所得的基因表达量数据,可预测所述植物的种子收量。即,即使关于检查对象的植物不经栽培试验,也可从可由第二代测序仪简易地取得的基因表达量数据,推测上述的种子收量等的目的变量。
如上所述,利用本实施方式中所示的转录组解析装置可对于指定的目的变量而制成预测模型式。通过使用制成的预测模型式,可例如如图8所示构建检查对象生物的特性评价系统20。
图8中所示的特性评价系统具备:储存关于用本实施方式中所示的转录组解析装置制成的指定的目的变量的预测模型式的存储部21,及基于关于检查对象生物的基因表达数据而预测目的变量的预测部22。存储部21针对每个检查对象的生物而对于各种各样的目的变量而储存预测模型式。例如存储部21可对于作为检查对象的植物,对于地上部重量、根部重量、叶面积、种子收量、高温度处理时间、低温度处理时间、药剂处理浓度、病害虫应激时间等的目的变量而储存各自预测模型式。
当预测部22被输入关于检查对象的生物的基因表达量数据时,可向储存在存储部21的预测模型式各自代入基因表达量数据,对于各种目的变量而算出预测值。
这样,特性评价系统20通过对于各种各样的目的变量而各自将预测模型式储存在存储部21,可基于检查对象生物的基因表达量数据而输出关于这些目的变量的预测值。例如,如果对于指定的植物输入基因表达量数据,则可对于地上部重量、根部重量、叶面积、种子收量、高温度处理时间、低温度处理时间、药剂处理浓度、病害虫应激时间等的目的变量而一并或以选择的范围得到预测值。
以上说明的第1实施方式及第2实施方式涉及的转录组解析装置及解析方法例如,可由具备中央处理装置(cpu)、主存储装置、辅助记忆装置、输出装置及输入装置的计算机实现。即,例如,目的变量数据及基因表达量数据可在中央处理装置的控制下,经输入装置而输入,可存储在主存储装置或辅助记忆装置。另外,例如,第m的子数据集可在中央处理装置的控制下、根据指定的算法而生成。再者,基于第m的子数据集的第m的预测式可在中央处理装置的控制下、根据指定的算法而生成。这样,以上说明的第1实施方式及第2实施方式涉及的转录组解析装置及解析方法可在中央处理装置的控制下实现。
但是,以上说明的第1实施方式及第2实施方式涉及的转录组解析装置及解析方法也可由所谓的云计算实现。在云计算中,例如,可利用储存在云服务器的目的变量数据及基因表达量数据,另外,也可将生成的预测式或基因列表储存在云服务器。
【实施例】
以下,由实施例更详细地说明本发明,但本发明的技术性范围不限于这些实施例。
〔实施例1〕
1.材料及方法
1-1.实验材料稻系统和栽培条件
在本实施例中,ouu365/arrozdaterra//ouu365回交自交系统(bils)使用在fukudaetal.,2014,plantproductionscience17:41-46.中记述的系统。将系统种子用稀释50倍次氯酸消毒,用自来水清洗3次之后,在30℃水中浸渍2天而使发芽。将每1系统24粒的发芽种子播种到水培养栽培用浮子上(fukudaetal.,2012,plantproductionscience15:183-191.),使在水培养栽培用溶液上生长(hayashiandchino,1986,plantandcellphysiology27:1387-1393.)。水培养液每隔2天制作更换,使在20℃、12小时明暗周期的生长室内生长14天。
1-2.方法
1-2-1.qtl解析
采集发芽14天后的bil104系统和亲本2系统的苗,用干燥机于80℃干燥2天之后,去除种子和根部分,秤量。实验由3次反复的生物学重复进行,在qtl解析中使用苗地上部干物重量的平均值。bil的基因型使用124种ssr标志物而进行解析(fukudaetal.,2014,plantproductionscience17:41-46.),使用mapmaker/exp3.0(landeretal.,1987,genomics1:174-181.doi:10.1016/0888-7543(87)90010-3)和qtlcartographer2.5(由wangetal.,2010,statisticalgenetics&bioinformatics,northcarolinastateuniversity提供)而进行qtl解析。
1-2-2.rna的单离和rna-seq
选出亲本品种的ouu365和arrozdaterra、以及各自初期生长量不同的bil20系统,在rna-seq解析中使用。对于发芽14天后的苗而去除种子和根部分,对苗地上部的生鲜重量进行测定之后,在液氮中冷冻,于-80℃保存至在解析中使用。使用rneasyminikit(qiagen公司制)而提取rna之后,进行rna-seq解析。将rna的定量-定性使用2100-bioanalyzer(agilenttechnologies公司制)而进行之后,使用truseqrnaltsamplepreparationkitv2(illuminainc公司制)而制成测序用库。由illuminahiseq2000,用100bp,单末端读长,进行库的测序。测序结果的fastq文件示于ddbjsequencereadarchive(dra)、登录号.dra006312。
测序数据以oryzasativa-nipponbare-reference-irgsp-1.0基因组(oryzasativa.irgsp-1.0.24.dna.toplevel.fa.gz,ftp://ftp.ensemblgenomes.org/pub/release-24/plants/fasta/oryza_sativa/dna/)及基因集(oryzasativa.irgsp-1.0.24.gtf.gz,ftp://ftp.ensemblgenomes.org/pub/release-24/plants/gtf/oryza_sativa/)作为参照序列,使用tophat2(kimetal.,2013,genomebiology14:13.doi:10.1186/gb-2013-14-4-r36;trapnelletal.,2009,bioinformatics25:1105-1111.doi:10.1093/bioinformatics/btp120)而进行定位。对于各基因的表达量,作为fpkm(fragmentsperkilobasemillion)值算出。
1-2-3.表达量生物标志物和基因选出频度的算出
对于表示苗地上部生鲜重的表达量生物标志物和基因的选出频度而用以下的方法算出。对于表达量的平均值是0.01以上的基因37043种而如下所述用于解析。对于各基因的表达量而向fpkm值加0.01之后变换为log2值。对于表达量生物标志物而使用lasso法,由l1线形回归模型进行选出(tibshirani,1996,journaloftheroyalstatisticalsocietyseriesb-methodological58:267-288.)。为了计算生物标志物基因的选出频度,重复进行使用转录组的部分集团(subset)的生物标志物的选出。从37043个基因之中随机地选择10%的基因,作为变量而在lasso解析中使用。以从输入的变量之中选出8个基因作为适合的,系数不是0的说明变量,作为表达量生物标志物。部分集团(subset)的选出和表达量生物标志物的算出重复进行10000次。将各基因的选出频度确定为在10000次的试验中用于生物标志物的比例。解析使用r的glmnetpackage(rcoreteam,2015,r:alanguageandenvironmentforstatisticalcomputing.https://www.r-project.org/)而进行。
1-2-4.sg1基因的测序
对于ouu365和arrozdaterra的sg1基因的编码区域、及上游-2108bp的区域,由pcr,使用以下的引物进行扩增:5’-gggacgtgataaccgactca-3’(seqidno:1)及5’-ccccactgtacgttctctcc-3’(seqidno:2)。将pcr产物使用illustraexoprostarkit而进行纯化,送到fasmac公司而进行测序。
为了对于翻译开始点起-1948bp上游的1碱基取代而进行检测,使用以下的引物而扩增pcr产物:5’-gggacgtgataaccgactca-3’(seqidno:3)及5’-ttcaggtcacctagcccatc-3’(seqidno:4)、由限制性内切酶haeiii进行切断。arrozdaterra类型的序列ggcc由haeiii切断,但ouu365类型的序列agcc不被切断。
1-2-5.定量实时pcr
从苗地上部,如上所述提取总rna。使用1μg的总rna而由primescriptrtreagentkitwithgdnaeraser(takarabio公司)进行cdna的合成。使用thermalcyclerdicerealtimesystemiii,由sybrpremixextaq和引物组oa045647(takarabio公司),将sg1cdna量的定量由实时pcr进行。实时pcr的测定由3次反复的生物学重复进行。为了sg1mrna的拷贝数的算出,以ouu365的cdna作为模板,将sg1的pcr产物使用以下的引物而扩增:5’-cgaccagctgatctccaa-g3’(seqidno:5)及5’-catttttactggcccttcca-3’(seqidno:6),作为实时定量pcr的标准品使用。标准品用pcr产物使用qubit荧光计(thermofisherscientific公司)而进行定量,从其分子量算出拷贝数。sg1表达量(copiesperngrna)变换为log2值之后,在qtl解析中使用。
2.结果
2-1.回交自交系统(bil)的qtl解析
arrozdaterra和ouu365的发芽14天后的地上部干物重的平均各自是5.11mg、2.91mg,arrozdaterra显著地重(t-检验,5%水平)。bil104系统的地上部干物重量分布于2.52~5.47mg之间(图9)。使用bil104系统而进行地上部干物重的qtl解析的结果,检测到在第3、7及10染色体上以arrozdaterra类型使地上部干物重增加的qtl(表1、图10)。再者,图10中,黑色四角形表示使地上部干物重增加的qtl的位置。空白椭圆表示使sg1表达量降低的eqtl的位置。
【表1】
2-2.rna-seq解析和生物标志物基因的选出
为了与初期生长量具有关联的转录产物的探索,使用亲本品种2品种和从bil之中具有不同的初期生长量的20系统(图11)、从发芽14天后的苗地上部提取rna,在rna-seq解析中使用。再者,图11中的空白色三角形对于在rna-seq分析中使用的bil系统各自而表示地上部干物重的平均值。苗地上部生鲜重示于图12。每个样品得到了平均41.6m的读长数,相当于96.1%的40.0m读长/样品在os-nipponbare-reference-irgsp-1.0genome上定位。基因表达量作为fpkm值(每百万个读长的编码序列的每千碱基的片段(fragmentsperkilobaseofcodingsequencepermillionreads))算出。对于成为表示苗地上部生鲜重的生物标志物的基因的选出频度而使用上述“1-2-3.表达量生物标志物和基因选出频度的算出”中所示的方法而如以下一样确定。从全表达基因之中随机地选择10%的基因而作为部分集团(subset),使用lasso解析而从部分集团内算出8个基因作为表示苗地上部生鲜重的说明变量(表达量生物标志物)。将部分集团(subset)的选出和表达量生物标志物的算出重复10000次,确定各基因作为表达量生物标志物被选出的频度。以高频度选出的基因显示其表达量与苗地上部生鲜重连动。以高频度(1%以上的确立)选出158个基因。这些选出的158个基因的列表示于图13。选出的158个基因的表达量全部与地上部生鲜重具有显著的相关(5%水平)。
2-3.苗地上部重qtl内所含的高频度选出生物标志物基因
当对选出的高频度基因和苗地上部重量qtl进行比较时,第3、7及10染色体上qtl内所含的基因各自有5个、6个及4个。这之中在第3染色体上基因之中,含既有的低温发芽基因、qltg3-1(rapid:os03g0103300,fujinoetal.,2008,theoreticalandappliedgenetics108:794-799.doi:10.1007/s00122-003-1509-4)。在rna-seq中使用的系统的qltg3-1表达量和地上部生鲜重之间见到显著的正的相关(图14)。亲本品种的一个arrozdaterra报告了有功能型的qltg3-1基因(fujinoandiwata,2011,theoreticalandappliedgenetics123:1089-1097.doi:10.1007/s00122-011-1650-4)、另一方的亲本品种ouu365在qltg3-1基因编码区域内具有71bp的缺损,确认到失其功能(fukudaetal.,2014,plantproductionscience17:41-46)。调查在rna-seq中使用的bil系统的qltg3-1基因型的结果,具有arrozdaterra类型的qltg3-1的系统的地上部生鲜重和qltg3-1表达量与具有ouu365类型的qltg3-1的系统相比,显著地高(t-test,1%level)。
2-4.处于苗地上部重qtl外的高频度选出生物标志物基因
在不含在苗地上部重量qtl内的高频度选出基因之中,含既有的组织延伸抑制基因sg1(shortgrain1,rapid:os09g0459200,nakagawaetal.,2012,plantphysiology158:1208-1219.doi:10.1104/pp.111.187567)。在rna-seq中使用的系统的sg1基因表达量和地上部生鲜重具有显著的负的相关(图15)。已知sg1在过量表达转化体中使植物激素向油菜类固醇的应答性降低,使植物体矮化(nakagawaetal.,2012,plantphysiology158:1208-1219.doi:10.1104/pp.111.187567)。但是,sg1是否具有自然变异至今未被报告。对亲本品种的arrozdaterra和ouu365的sg1基因的碱基序列进行比较的结果,在编码区域内无碱基取代或缺失-插入变异。在翻译开始点的上游-1948b和-2038b的位置有单碱基取代,但在rna-seq解析中使用的系统的sg1基因表达量由此位置的基因型见不到差异。
2-5.bil104系统的sg1表达量的定量实时pcr解析
为了确认在rna-seq解析中使用的以外的bil系统中sg1表达量和苗地上部重量是否也具有相关,对于bil104系统全部和亲本品种而进行利用定量实时pcr的sg1表达量的测定。结果,bil104系统和亲本品种的sg1表达量与地上部生鲜重显示显著的负的相关(图16)。未见到翻译开始点的上游-1948b的基因型导致的sg1的表达量的差异。为了调查影响sg1表达量的染色体区域,使用bil104系统的sg1表达量而进行表达量qtl解析(eqtl解析)的结果,在第3染色体上和第7染色体上这2处检测到以arrozdaterra类型降低sg1表达量的eqtl(表2及图10)。其中,作用力强的第7染色体上的eqtl与苗重量qtl处于同位置(图10)。一方面,在存在sg1基因的第9染色体上检测不到eqtl。
【表2】
由本实施例得知,可由使用bil20系统及亲系统的rna-seq解析选出成为表示初期生长的指标的生物标志物基因候选。另外,在其中,含既有的组织延伸抑制基因sg1。sg1具有组织延伸抑制的作用由利用活化标签(activation-tag)的过量表达转化体确认(nakagawaetal.,2012,plantphysiology158:1208-1219.doi:10.1104/pp.111.187567),但在自然状态下,sg1表达量在系统间是否有差异不明。由本实施例的转录组解析得知,bil系统的sg1表达量和苗地上部生鲜重具有负的相关(图15)。再者,由定量实时pcr解析得知,不仅是在rna-seq中使用的系统,在104的bil系统全部中,sg1表达量和苗地上部生鲜重也具有负的相关(图16),提示sg1影响苗的初期生长量。从这些结果认为,使用22系统的rna-seq数据的,本实施例的转录组解析是对于检测关于初期生长的转录产物而言有效的手段。
2-6.本实施例中所示的转录组解析的有用性
转录产物的整体解析(转录组解析)是可检测影响各种各样的形态-生理学性质的转录产物的强力的手段,而转录产物复杂地受多个环境要因-遗传要因的影响。因此,为了统计学地选出表示特定的性质的表达量生物标志物,认为期望使用数百以上的多数的样品数以去除噪声。但是,准备数百以上的多数的样品数,进行rna-seq等的基因表达解析多有困难的情况。
在本实施例中所示的转录组解析中,使用称为bil20系统及亲本品种2系统的22系统的比较小的样品尺寸而试了表示苗重量的表达量生物标志物的检测。结果,由本实施例中所示的转录组解析,作为候选生物标志物,检测到具有和不具有qltg3-1和sg1这样的基因组变异的2种既有的基因。结果显示,本实施例中所示的转录组解析即使是比较小的样品尺寸的解析,也可有效进行表达量生物标志物的选出。
〔实施例2〕
在本实施例中,使用在实施例1中制成的高频度基因列表(图13)而算出苗地上部生鲜重的预测值。
1.方法
使用在实施例1中制成的高频度基因列表(图13)158个基因之中上位100个基因的基因表达量及苗地上部生鲜重(图12),由随机森林法(breiman,l.,2001,machinelearning45:5-32),从基因表达量预测苗地上部生鲜重。在随机森林中,以关于这100个基因的在实施例1中测定的表达量数据和苗地上部生鲜重量作为输入值,以决策树的形式制成预测模型式,基于所述预测模型式而从关于上述100个基因的表达量数据算出预测值。
2.结果
将5折交叉验证(crossvalidation)重复20次,求出苗地上部生鲜重的预测值。以横轴作为苗地上部生鲜重的实测值,作为将纵轴由上述预测模型式算出的预测值(平均值)标绘数据的坐标图示于图17。当对于图17中所示的数据而算出r2(自由度调整济决定系数)时成为0.8554,显示非常高的拟合度。即,表示使用关于在实施例1中制成的列表中所含的基因的基因表达量数据及苗地上部生鲜重而策划的预测模型式适用于实际的数据,可认为说明变量(基因表达量数据)良好地说明目的变量(苗地上部生鲜重)。
序列表
<110>toyotajidoshakabushikikaisha
theuniversityoftokyo
nationalagricultureandfoodresearchorganization
ryukokuuniversity
<120>转录组解析装置及解析方法
<130>ph-7609-us
<150>jp2018-3697
<151>2018-1-12
<160>6
<170>patentinversion3.5
<210>1
<211>20
<212>dna
<213>人工序列
<220>
<223>合成的dna
<400>1
gggacgtgataaccgactca20
<210>2
<211>20
<212>dna
<213>人工序列
<220>
<223>合成的dna
<400>2
ccccactgtacgttctctcc20
<210>3
<211>20
<212>dna
<213>人工序列
<220>
<223>合成的dna
<400>3
gggacgtgataaccgactca20
<210>4
<211>20
<212>dna
<213>人工序列
<220>
<223>合成的dna
<400>4
ttcaggtcacctagcccatc20
<210>5
<211>19
<212>dna
<213>人工序列
<220>
<223>合成的dna
<400>5
cgaccagctgatctccaag19
<210>6
<211>20
<212>dna
<213>人工序列
<220>
<223>合成的dna
<400>6
catttttactggcccttcca20
- 还没有人留言评论。精彩留言会获得点赞!