一种抑制阿片类镇痛药物不良反应的制剂及其应用的制作方法
本发明涉及医药领域,具体涉及一种抑制阿片类镇痛药物不良反应的制剂及其应用。
背景技术:
癌症病人受到病痛折磨,临床上一般采用阿片类镇痛药物进行镇痛治疗,缓解癌症病人疼痛感受,但是阿片类药物容易产生耐受性和依赖性。
耐受性,即镇痛效力的丧失,是阿片类药物治疗的常见并发症之一,导致剂量需求不断增加并且随着时间的推移而降低有效性。
依赖性,是由涉及自主神经和躯体活动过度的阿片类药物戒断综合征所揭示的改变的生理状态的发展。
阿片类药物的耐受性和依赖性是众所周知的生理现象,大量研究发现,阿片类药物不良反应包括耐受、依赖的机制相当复杂,涉及到兴奋性氨基酸、5-羟色胺、多巴胺、去甲肾上腺素和乙酰胆碱等多种神经递质。目前,临床上除了采用阿片受体拮抗剂外,没有明显抑制阿片依赖和耐受的药物,但是使用阿片受体拮抗剂的副作用也很明显,就是在抑制阿片依赖耐受的同时,阿片的镇痛镇静作用也被完全抑制。国际医药业界对阿片类药物不良反应的发生和抑制技术研究也比较多,比如由日本协和发酵麒麟株式会社申请的专利号为200980119667.0的“镇痛耐受性抑制剂”,目的都是开发能够抑制阿片类药物不良反应的制剂。
抑制阿片类镇痛药物的耐受性和依赖性尤其重要,因为阿片类镇痛药物是缓解癌症病人,特别是晚期癌症病人癌痛的重要药物,随着用药时间的延长,阿片类镇痛药物的剂量越来越大,效果越来越差,并且导致病人产生依赖性,对晚期癌症病人来说,无异于雪上加霜。因此,开发一种能够抑制阿片类镇痛药物的耐受性和依赖性的制剂,使其能在需要长时间镇痛的病人治疗中得以应用是非常重要的。
技术实现要素:
本发明的目的在于提供一种抑制阿片类镇痛药物不良反应的制剂,解决上述背景技术中提出的问题。
为实现上述目的,本发明提供如下技术方案:
一种抑制阿片类镇痛药物不良反应的制剂,所述制剂中含有17-n-烯丙基氨基-17-去甲氧基格尔德霉素或/和其同系物的一种或几种。
进一步,所述17-n-烯丙基氨基-17-去甲氧基格尔德霉素及其同系物的分子式如下(i)~(vi)所示:
其中:(i)中,当r为och3时,(i)为格尔德霉素;
当r为nhch2ch=ch2时,(i)为17-n-烯丙基氨基-17-去甲氧基格尔德霉素,简写为17-aag;
当r为nhch2ch2n(ch3)2时,(i)为17-脱甲氧基-17-[[2-(二甲基氨基)乙基]氨基]格尔德霉素,简写为17dmag。
优选地,所述制剂中含有17-aag。
进一步,所述阿片类镇痛药物是作用于μ阿片受体而发挥镇痛作用的药物。
进一步,所述阿片类镇痛药物包括:盐酸吗啡片、硫酸吗啡缓释片、磷酸可待因片、盐酸吗啡注射液、盐酸哌替啶注射液、芬太尼注射液其中的一种或几种的组合。
具体的,所述制剂的效果包括降低癌症患者使用阿片类药物时产生的镇痛耐受性、减少对阿片药物依赖。
优选地,所述17-aag或/和其同系物的有效量为经脑室给药的药物中含量为0.1-1000nmol。
优选地,所述17-aag或/和其同系物的有效量为经腹部给药的药物中含量为0.1-1000nmol。
所述17-aag或/和其同系物在癌症患者使用的抑制阿片类镇痛药物不良反应的药物中的应用。
本发明的有益效果是:
1、17-aag伴随吗啡同时给药,急性给药时会抑制吗啡的镇痛效果,但慢性给药时能明显抑制吗啡的耐受性:从实施例2可以看出,17-aag伴随吗啡同时给药,慢性给药7天,实验小鼠对吗啡镇痛作用的抑制(耐受作用)斜率明显降低,给药7天后,用纳洛酮(10mg/kg)催促戒断,17-aag伴随吗啡慢性给药能够明显抑制催促戒断反应症状。
2、17-aag伴随吗啡同时给药,能显著抑制吗啡的躯体耐受和精神依赖性:实施例3可以看出,吗啡联合17-aag药物组小鼠在条件性训练中不能形成条件性位置偏爱,而吗啡联合溶媒组和吗啡组皆能形成明显的条件性位置偏爱,说明17-aag不仅能够明显抑制阿片类药物躯体耐受和依赖,也能够明显抑制阿片类药物的精神依赖。
3、17-aag及其同系物同时具有抑制肿瘤细胞生长和抑制阿片类镇痛药物不良反应的特性,可以减少癌症病人治疗成本和多种药物混合使用导致的不良反应:μ-阿片受体(mor)在阿片类物质诱导的镇痛、耐受及依赖性等过程中发挥着重要的作用。热休克蛋白90(hsp90)是癌症治疗中的有希望的靶标,肿瘤细胞对hsp90有明显的依赖性,hsp90在肿瘤细胞中含量是正常细胞的2~3倍,hsp90对mor磷酸化有增强作用。目前有研究认为,mor磷酸化受体内吞可能是阿片类药物依赖耐受的生物学机制之一,而本发明发现,17-aag能够明显抑制hsp90β对mor磷酸化的增强作用(实施例4)、能够明显抑制hsp90β对细胞外信号调节激酶(erk)磷酸化的增强作用(实施例5)。由于癌症患者普遍需要长期使用阿片类药物镇痛,会产生不良反应,特别是依赖和耐受,影响了阿片类药物的镇痛效果,因此又需要同时服用抑制阿片类镇痛药物不良反应的药物。而在本发明的技术方案下,通过合理地使用17-aag及其同系物和阿片类镇痛药物,同时具有抗肿瘤和抑制阿片类镇痛药物耐受性和依赖性的双重作用,进而可以减少其他药物的使用。
附图说明
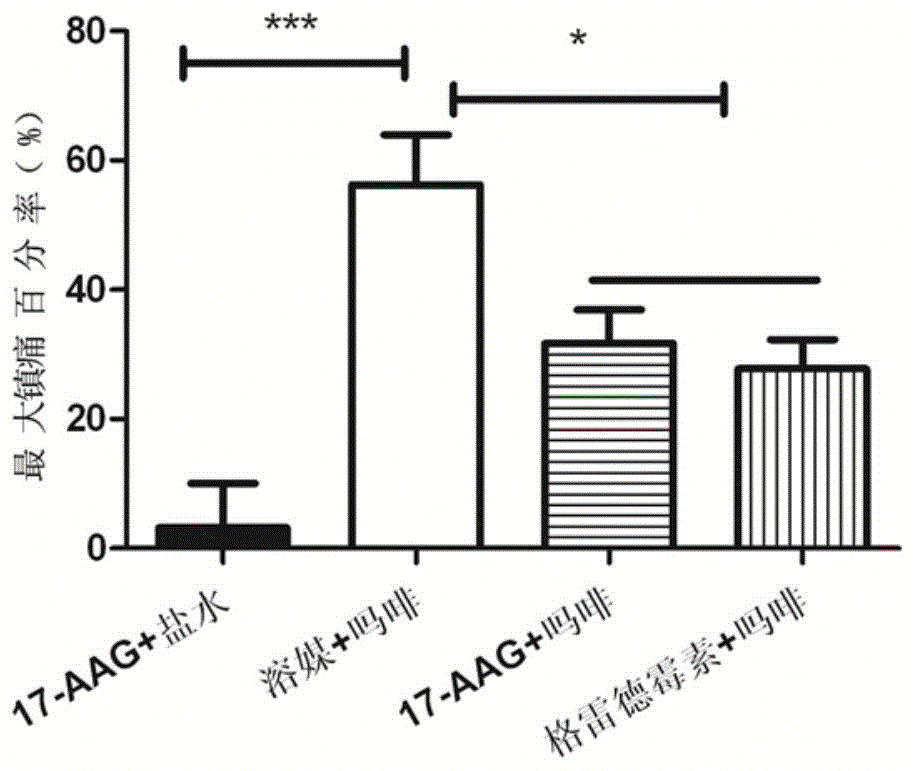
图117-aag和格尔德霉素急性给药时对吗啡镇痛作用的影响
图217-aag慢性给药时对吗啡镇痛率的影响
图317-aag慢性给药时对吗啡镇痛耐受性的影响
图417-aag慢性给药时对吗啡戒断反应的影响
图517-aag慢性给药对吗啡条件性位置偏爱的实验时间设计
图617-aag慢性给药对吗啡条件性位置偏爱的评分
图717-aag慢性给药对吗啡条件性位置偏爱的伴药箱运动距离
图8hsp90β能够明显促进mor磷酸化的电泳图
图917-aag能够明显抑制hsp90β对mor磷酸化的增强作用的电泳图
图10格尔德霉素也能够明显抑制hsp90β对mor磷酸化的增强作用的电泳图
图11hsp90β能够明显促进erk磷酸化的电泳图
图1217-aag能够明显抑制hsp90β对erk磷酸化的增强作用的电泳图
图13格尔德霉素也能够明显抑制hsp90β对erk磷酸化的增强作用的电泳图
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1:17-aag伴随吗啡同时给药,急性给药的实验及结果
c57/b6小鼠共40只,随机分为4组,在给药前热板(55℃)测定动物基础痛域,随后分别脑室给药5微升的17-aag(10nmol)、溶媒(solvent)、17-aag(10nmol)和格尔德霉素(10nmol),15分钟后,依次皮下给药生理盐水(10ml/kg)、吗啡(mor,10mg/kg)、吗啡(mor,10mg/kg)、吗啡(mor,10mg/kg),给药后30min热板测动物痛域。最大镇痛百分率(%mpe)=(给药后痛域-给药前痛域)/(60-给药前痛域)×100%。从表1和图1中可见17-aag对小鼠没有镇痛作用,吗啡对小鼠有明显的镇痛作用,17-aag(10nmol)和格尔德霉素(10nmol)能够明显抑制吗啡的镇痛作用。
表117-aag和格尔德霉素急性给药时对吗啡镇痛作用的影响
注:n=10,平均值±标准误,***p<0.001,与17-aag(10nmol)+盐水组比较;#p<0.05,与溶媒+吗啡(10mg/kg)比较。
此外,在其他品系的小鼠(昆明、icr),也可以观察到17-aag和其同系物格尔德霉素(geldanamycin)无论是脑室给药还是腹腔给药,都能明显抑制吗啡急性给药引起的镇痛作用。
实施例217-aag伴随吗啡同时给药,慢性给药的实验及结果
c57/b6小鼠共30只,在实验环境中饲养2-3天,随机分为3组,每天给药两次,上午8:00点开始,在给药前热板(55℃)测定动物基础痛域,随后分别脑室给药5微升的溶媒(solvent)、17-aag(10nmol),15min后皮下给药吗啡(mor,100mg/kg),吗啡给药30min测痛域。下午6:003组动物只给吗啡(100mg/kg),连续给药及测痛域7天,末次给药后2小时纳洛酮(10mg/kg)腹腔注射给药,给药后立即放入戒断观察盒内,观察小鼠在15min内的跳跃、扭体、甩头、震颤、理毛等戒断症状(withdrawlsigns)并进行评分。从表2和图2可见,吗啡(100mg/kg)在第一天给药后,镇痛百分率高达94%,而17aag(10nmol)能够明显抑制吗啡的镇痛作用,使镇痛率降至78.6%,但是溶媒脑室给药后对吗啡的镇痛作用没有明显影响,镇痛率为98%。但随着给药时间的延长,吗啡的镇痛作用出现下降,即吗啡耐受,到第7天镇痛率为33.69%,而17aag能够明显抑制吗啡的耐受作用,到第7天镇痛率为51.16%,从图3可见,17aag+吗啡组的镇痛下降的斜率明显低于溶媒+吗啡组和单用吗啡组。从表3和图4可见,采用纳洛酮催促戒断后,17aag能够明显抑制吗啡躯体依赖戒断症状。
表217-aag慢性给药时对吗啡镇痛耐受性的影响
注:***p<0.001,**p<0.01,*p<0.05与同组第一天比,#p<0.05,17-aag预处理组与溶媒预处理或单用吗啡组比较,双因素方差分析。
表317-aag慢性给药时对吗啡戒断反应的评分
注:#p<0.05,17-aag预处理组与溶媒预处理或单用吗啡组比较,单因素方差分析。
综上可见,在吗啡慢性给药躯体依赖催促戒断模型上,吗啡联合17-aag药物组在给药后第2到第7天,较吗啡联合溶媒组镇痛作用明显增强,并且17-aag能够明显抑制吗啡的躯体依赖和耐受。17-aag抑制吗啡的耐受和依赖的明确的分子生物学及神经环路机制还有待于进一步研究,但有可能是这些递质在不同脑区都受到紧张性抑制,维持正常的递质平衡,在阿片类药物作用于中枢或外周的mor后,抑制了gaba能中间神经元,使v-氨基丁酸释放减少,下游递质释放脱抑制后,导致一些不良反应的发生。
实施例317-aag伴随吗啡同时给药,对精神依赖性影响的实验和结果
17-aag慢性给药对吗啡条件性位置偏爱的实验时间设计见图5,采用c57/b6小鼠共50只,动物在实验环境中饲养2-3天,预适应期(d1-d3):3天,每天适应1次,每次15min,记录后两次15min内两侧停留时间,以d2、d3两次平均值作为前测结果,筛除不合格动物。
动物剔除标准:总体动物在黑箱和白箱两侧箱体中的停留时间之差超过100s,认为存在自然偏性,将两侧箱体中停留时间接近的个体予以剔除;总体动物在两侧箱体中的停留时间之差不超过100s,认为不存在自然偏性,将任意一侧箱体中停留时间超过560s的个体予以剔除。基本状态差的个体予以剔除,每次预适应取出后皮下注射盐水,使动物熟悉操作。
条件性训练期(d4-d8):根据前测结果,选取40只动物随机分为4组,每组10只。选择偏性或非偏性程序,如果是偏性,在非偏爱侧伴药;如果是非偏性,采用抗衡设计,即一半动物在黑箱伴药,另一半在白箱伴药。每天上午8:00开始和下午14:30开始各训练一次,45min/次,连续训练5个周期(5天)。插上隔板,使动物限制在黑箱或白箱。每只动物每天接受两次任务训练,30min/次;交替给予灭菌注射用水或相应药物进行训练,训练次序先后平衡。具体过程为:每组的动物上午给予灭菌注射用水后非伴药侧训练,下午给予相应药物后伴药侧训练。以此类推,连续训练5天。每天上午8:00-12:30训练,下午14:30-19:00训练,两次任务训练之间至少间隔4~6h。吗啡10mg/kg与生理盐水的试验组为皮下给药,17aag(10nmol/5μl)和溶媒(5μl)的试验组为脑室给药。
测试期(d9):训练期结束后第二天进行测试,动物不给药,记录15min内伴药侧和非伴药侧停留时间。小鼠在白箱停留时间-黑箱停留时间即为条件性位置偏爱得分。
表417-aag对吗啡条件性位置偏爱的影响
注:#p<0.05,17-aag预处理组与溶媒预处理吗啡组比较,单因素方差分析。
从表4和图6和图7可见,从伴药箱运动距离和条件性位置偏爱得分上开看,在小鼠的条件性位置偏爱模型(cpp)上,吗啡联合17-aag药物组小鼠在条件性训练中不能形成有效的条件性位置偏爱,而吗啡联合溶媒组能形成明显的条件性位置偏爱。说明17-aag不仅能够明显抑制阿片类药物躯体依赖,也能够明显抑制阿片类药物的精神依赖。17-aag及其类似物正在进行抗肿瘤的ii期临床试验,阿片类药物又是常用的癌症镇痛药,所以在两药联合用药时,17-aag及其类似物会明显减低阿片类药物的依赖、耐受与成瘾。当然,在非癌性镇痛,需要阿片类药物长期用药时,17-aag及其类似物也能发挥同样的作用。
实施例4:用17-aag抑制hsp90蛋白增强μ-阿片受体(mor)磷酸化效果的实验和结果
本实验采用mor-cho用脂质体法瞬时转染pcdna3.1myc/hisb-hsp90β或pcdna3.1myc/hisb(vector)后24h,给予17-aag(10μm)或溶媒(solvent)作用20h,最后给予mor受体激动剂damgo(10μm),分别作用于细胞5,15,30min。用ripa细胞裂解液在冰上裂解细胞30min。14000rpm(转/分钟)离心15min后,收集细胞上清液,加入5×loadingbuffer,沸水变性5min,蛋白用10%sds-page凝胶100v电泳90min后,160ma转印至pvdf膜,用5%脱脂奶粉封闭后用分别用抗mor磷酸化抗体(1∶1000)检测mor磷酸化,用抗-mor抗体(1∶1000)检测mor,用抗myc抗体(1∶2000)检测hsp90β。
从图8可见hsp90β能够明显促进mor磷酸化,表现为mor磷酸化蛋白条带明显加深。而在给予17-aag(10μm)作用20小时后,与溶媒(solvent)比较,能够明显抑制hsp90β对mor磷酸化的增强作用,蛋白条带明显变浅(图9)。同样,格尔德霉素(geldanamycin,10μm)作用20小时后,也能够明显抑制hsp90β对mor磷酸化的增强作用(图10)。17-aag及其类似物能够抑制m0r的磷酸化,可能是17-aag及其类似物能够抑制阿片类药物的镇痛耐受及依赖等不良反应的分子机制。
实施例5:用17-aag抑制hsp90蛋白增强erk磷酸化效果的实验和结果
本实验采用mor-ch0脂质体法瞬时转染pcdna3.1myc/hisb-hsp90β或pcdna3.1myc/hisb(vector)后24h,给予17-aag(10μm)或溶媒(solvent)作用20h,最后给予mor受体激动剂damgo(10μm),分别作用于细胞5,15,30min.用ripa细胞裂解液在冰上裂解细胞30min。14000rpm(转/分钟)离心15min后,收集细胞上清液,加入5×loadingbuffer,沸水变性5min,蛋白用10%sds-page凝胶100v电泳90min后,160ma转印至pvdf膜,用5%脱脂奶粉封闭后用分别用抗erk磷酸化抗体(1∶1000)检测erk磷酸化,用抗-erk抗体(1∶1000)检测erk,用抗myc抗体(1∶2000)检测hsp90β。
从图11可见hsp90β能够明显促进erk磷酸化,表现为erk磷酸化蛋白条带明显变深。而在给予17-aag(10μm)作用20小时后,与溶媒(solvent)比较,能够明显抑制hsp90β对erk磷酸化的增强作用,表现为erk磷酸化蛋白条带明显变浅(图12)。同样,格尔德霉素(geldanamycin,10μm)作用20小时后,也能够明显抑制hsp90β对erk磷酸化的增强作用(图13)。在激动剂作用后,mor磷酸化受体内吞进而招募β-arresting,激活下游的mapk通路,使得erk磷酸化上调,这是是阿片类药物依赖耐受的生物学机制之一,17-aag及其类似物能够抑制erk的磷酸化,有可能就是17-aag及其类似物能够抑制阿片类药物的镇痛耐受及精神依赖行为的分子机制。
对于本领域技术人员而言,显然本发明不限于上述示范性实施例的细节,而且在不背离本发明的精神或基本特征的情况下,能够以其他的具体形式实现本发明。因此,无论从哪一点来看,均应将实施例看作是示范性的,而且是非限制性的,本发明的范围由所附权利要求而不是上述说明限定,因此旨在将落在权利要求的等同要件的含义和范围内的所有变化囊括在本发明内。不应将权利要求中的任何附图标记视为限制所涉及的权利要求。
此外,应当理解,虽然本说明书按照实施方式加以描述,但并非每个实施方式仅包含一个独立的技术方案,说明书的这种叙述方式仅仅是为清楚起见,本领域技术人员应当将说明书作为一个整体,各实施例中的技术方案也可以经适当组合,形成本领域技术人员可以理解的其他实施方式。
- 还没有人留言评论。精彩留言会获得点赞!