一种检测肿瘤异质性程度的方法及系统与流程
本发明涉及生物信息学技术领域,特别涉及一种检测肿瘤异质性程度的方法及系统。
背景技术:
癌症是全球最主要的非传染性疾病之一,也是死亡率很高的一种病种,在我国,每年有接近430万人被诊断为癌症,有超过280万人死于癌症。
抗肿瘤靶向药物和免疫检查点抑制剂是目前治疗癌症较为有效的手段,目前比较公认的免疫检查点抑制剂anti-pd-(l)1疗效评估潜在指标如tmb(肿瘤突变负荷)、msi(微卫星不稳定)等都不能完全将免疫检查点抑制剂获益的患者有效筛选出来。最近有研究提出ith(肿瘤异质性)有可能作为新的免疫检查点抑制剂pd-1疗效评估潜在指标。ith是指肿瘤在生长过程中,经过多次分裂增殖,其子细胞呈现出分子生物学或基因方面的改变,从而使肿瘤的生长速度、侵袭能力、对药物的敏感性、预后等各方面产生差异。它是恶性肿瘤的特征之一。一般临床上建议这些免疫检查点抑制剂药物在用于肿瘤治疗前进行基因检测以确定是否适合用药,以及用何种药物。
目前该领域内并未有权威的量化和计算ith方法,这些方法计算出来的ith指标不能或者只能在少量数据集中验证出能够评估免疫检查点抑制剂anti-pd-(l)1的疗效。
例如,申请公布号为cn106676178a的中国专利公开了一种评估肿瘤异质性的方法及装置,其方法包括:1)对患者的cfdna进行测序(优选高通量测序),获得测序信息;2)利用测序信息确定ctdna变异,根据测序信息和确定的ctdna变异,计算变异等位频率,确定变异所在区域的实际总拷贝数,计算ctdna占cfdna的比例;3)根据步骤2)中确定的比例以及ctdna变异的测序信息和拷贝数信息对ctdna变异进行聚类,聚类得到的每一个簇确定为一个分子克隆,得到聚类的克隆层级;4)根据患者的克隆层级对其肿瘤异质性进行评估,患者的克隆层级越多,其肿瘤异质性越高。其主要缺陷在于,ctdna在血液中的含量较低,约占整个cfdna的1%甚至0.01%([1]diehlf,schmidtk,chotima,etal.circulatingmutantdnatoassesstumordynamics[j].naturemedicine,2008,14(9):985.[2]diehlf,lim,dressmand,etal.detectionandquantificationofmutationsintheplasmaofpatientswithcolorectaltumors[j].proceedingsofthenationalacademyofsciences,2005,102(45):16368-16373.),检测难度较大,测序成本较高;其次,该发明需要使用pyclone软件对ctdna变异进行聚类,因为ctdna的含量低,导致每个ctdna变异所在区域的实际总拷贝数检测的准确性降低(在1%ctdna含量中需要100个拷贝数以上的突变才能够被全基因组测序检测出,而且探针捕获测序无法检测)(molpariab,nichanie,torkamania.assessmentofcirculatingcopynumbervariantdetectionforcancerscreening[j].plosone,2017,12(7).)。此外,在其实施例中,每个患者检测出的ctdna突变数量较少,为2-8个不等,与实际应用的情况相符(abboshc,birkbaknj,wilsonga,etal.phylogeneticctdnaanalysisdepictsearly-stagelungcancerevolution[j].nature,2017,545(7655):446-451.),当突变数量较少时,pyclone软件计算的聚类克隆层级数量受突变数量的影响较大,ctdna突变数量多的患者对应的聚类克隆层级数更多,因此通过该方法计算出的克隆层级有可能更多地是反映患者的ctdna突变数量而不是肿瘤异质性,并且ctdna突变的数量也是与患者的生存期存在显著相关性(ocanaa,díez-gonzálezl,garcía-olmodc,etal.circulatingdnaandsurvivalinsolidtumors[j].cancerepidemiologyandpreventionbiomarkers,2016,25(2):399-406.)。
因此,现有技术无法对肿瘤异质性程度进行检测。
技术实现要素:
本发明主要解决的技术问题是免疫检查点抑制剂anti-pd-(l)1疗效评估缺乏足够的潜在指标问题,以及如何使ith指标能够在更大规模更大范围内对免疫检查点抑制剂pd-1的疗效进行评估。
根据第一方面,一种实施例中提供一种检测肿瘤异质性程度的方法,包括以下步骤:
获取来自于每一受试者的同一肿瘤组织样本和对照样本的测序数据,对所述肿瘤组织样本和对照样本进行体细胞变异检测,获得体细胞变异位点;
将所述体细胞变异位点按簇进行聚类;
根据聚类结果,如果肿瘤细胞簇比例最高的簇包含两个或两个以上变异的异常结果,则将肿瘤细胞簇比例最高的簇判断为主克隆突变簇,剩余的突变簇判断为亚克隆突变簇;如果肿瘤细胞簇比例最高的簇仅包含一个变异的异常结果,则将肿瘤细胞簇比例最高和次高的突变簇同时判断为主克隆突变簇,剩余的突变簇判断为亚克隆突变簇;
计算所述肿瘤组织样本中亚克隆突变簇中的体细胞变异数量占所有体细胞变异数量的比值,该比值即为肿瘤异质性数值。
本领域技术人员可以理解,体细胞变异也可称为体细胞突变,变异位点也可称为突变位点。
本领域技术人员可以理解,同一肿瘤组织样本和对照样本是指来源于同一受试者的肿瘤组织样本和对照样本。
在一些实施方案中,根据所述体细胞变异位点,计算变异等位测序深度、变异等位频率、变异位点拷贝数、肿瘤纯度值,根据所述体细胞变异位点以及变异等位测序深度、变异等位频率、变异位点拷贝数、肿瘤纯度值进行聚类分析,将所述体细胞变异位点按簇进行聚类。
在一些实施方案中,所述变异等位测序深度vi是指测序数据中在相应位点发生体细胞变异的变异序列的条数;
所述变异等位频率
所述肿瘤纯度值是指肿瘤细胞数量在所述肿瘤组织样本细胞总数中的占比pur,取值范围为(0,1],所述肿瘤细胞是指发生体细胞变异的所有细胞的总和;
所述变异位点拷贝数的计算过程如下:根据体细胞变异位点vari所在区域的拷贝数变异cnvi,计算所述体细胞变异位点vari所在区域的参考拷贝数ncni和实际总拷贝数ncni,其中:
并获得体细胞变异位点vari所在的两条染色体上等位特异的拷贝数变异cnvi,major、cnvi,minor,其中cnvi,major≥cnvi,minor,
从而计算出实际的等位特异的拷贝数cni,major、cni,minor:
在一些实施方案中,聚类分析时,对于任一类型的体细胞变异,将所述受试者的肿瘤组织样本中的细胞被分为三类:正常细胞(n)、不携带该变异的肿瘤细胞(twt)和携带该变异的肿瘤细胞(tmut),携带所述体细胞变异的肿瘤细胞(tmut))占所有肿瘤细胞(tmut+twt)的比例称为该变异位点的肿瘤细胞比例,如果两个或以上变异位点的变异肿瘤细胞比例满足在同一个分布模型中,则对所述同一个分布模型中的变异赋予相同的簇标签,聚类成一簇,称为一个克隆;
每个受试者的每个簇标签cj(j=1,…,c),都有与之对应的肿瘤细胞簇比例
如果所述肿瘤细胞簇比例
其中,j=1,...,c,k=1,...,c且k≠j;
同时将剩余的簇一同判断为亚克隆突变簇csub:
csub=cl,l∈{1,...,c},l≠j,l≠k;
统计主克隆突变簇cmain中体细胞变异位点vari的数量nmain和亚克隆突变簇csub中体细胞变异位点vari的数量nsub,计算肿瘤异质性数值ith,所述肿瘤异质性数值ith是指亚克隆突变簇中的体细胞变异位点数量占所有体细胞变异位点数量的比值:
本领域技术人员可以理解,体细胞变异位点vari也可被称为变异vari。
在一些实施例中,还包括:根据所述肿瘤组织样本中亚克隆突变簇中的体细胞变异数量占所有体细胞变异数量的比值设定阈值,将小于或等于所述阈值的样本所对应的受试者判定为低风险受试者,将大于所述阈值的样本所对应的病例判定为高风险受试者。
在一些实施例中,取所有受试者的肿瘤异质性数值的中位数作为判断每位受试者肿瘤异质性高/低的阈值,克隆层级低于该阈值的受试者,其肿瘤异质性较低,反之,则肿瘤异质性较高。
在一些实施例中,所述受试者为实体瘤患者,优选为肺癌、鼻咽癌或黑色素瘤患者。
在一些实施例中,所述体细胞变异选自点突变(snv)、插入/缺失(indel)、结构变异(sv)、拷贝数变异(cnv)中的至少一种。例如,在一些实施例中,具体可以为snv、indel,在另一些实施例中,还可以为snv、indel、sv,在另一些实施例中,还可以为snv、indel、sv、cnv。
在一些实施例中,所述肿瘤组织样本和对照样本的测序方法为全基因组测序、全外显子组测序或探针捕获测序,优选为全外显子组测序。
根据第二方面,提供一种检测肿瘤异质性程度的系统,所述系统包括:
数据获取模块,用于获取来自于每一受试者的同一肿瘤组织样本和对照样本的测序数据;
体细胞变异检测模块,用于对所述肿瘤组织样本和对照样本进行体细胞变异检测,获得体细胞变异位点;
聚类模块,用于将所述体细胞变异位点按簇进行聚类;
主克隆亚克隆判断模块,用于根据聚类结果,如果肿瘤细胞簇比例最高的簇包含两个或两个以上突变的异常结果,则将肿瘤细胞簇比例最高的簇判断为主克隆突变簇,剩余的突变簇判断为亚克隆突变簇;如果肿瘤细胞簇比例最高的簇仅包含一个变异的异常结果,则将肿瘤细胞簇比例最高和次高的突变簇同时判断为主克隆突变簇,剩余的突变簇判断为亚克隆突变簇;
肿瘤异质性程度计算模块:用于计算所述肿瘤组织样本中亚克隆突变簇中的体细胞变异数量占所有体细胞变异数量的比值,该比值即为肿瘤异质性数值。
根据第三方面,提供一种检测肿瘤异质性程度的装置,所述装置包括:
存储器,用于存储程序;
处理器,用于通过执行所述存储器存储的程序以实现如第一方面所述的方法。
根据第四方面,提供一种计算机可读存储介质,包括程序,所述程序能够被处理器执行以实现如第一方面所述的方法。
采用的变异检测软件包括但不限于varscan、mutect,具体可以为varscan(v2.4.1)、mutect(v4.0.12.0)等。
在一些实施方案中,采用的拷贝数检测软件包括但不限于cnvkit、ascatngs,具体可以为等cnvkit(v0.8.1)、ascatngs(v3.1.0)。
在一些实施方案中,聚类软件选自pyclone软件。
在一些实施例中,可以采用pyclone软件的其它版本或其它变异聚类分析软件,例如clonesig(v0.1)。
依据上述实施例的方法及系统,本发明提供了一种计算样本的肿瘤异质性程度的具体方法,充分利用样本各突变位点检测的突变频率估算出对应肿瘤克隆的细胞占比,从而推算出肿瘤异质性程度的具体数值,为后续预测免疫治疗疗效提供了一个数值上可以参考的依据。
附图说明
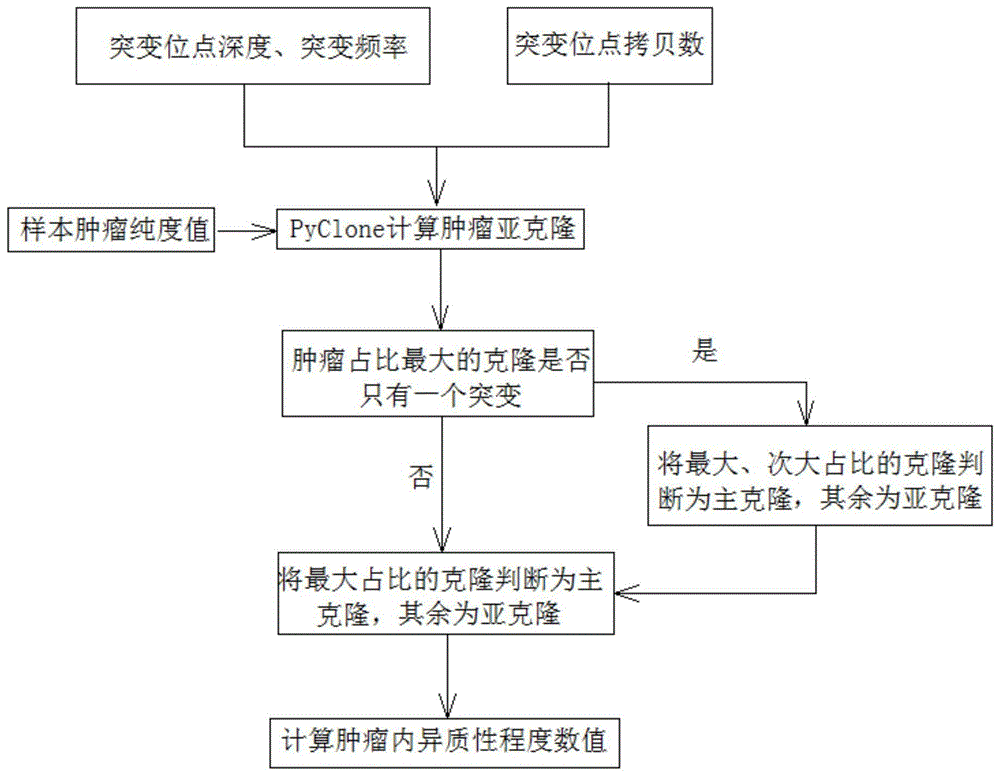
图1显示为本发明实施例中肿瘤异质性程度检测的流程框图;
图2显示为本发明实施例1中肺癌队列组织样本肿瘤异质性程度检测结果。
图3显示为本发明实施例1中肺癌队列组织样本肿瘤异质性程度预测免疫治疗疗效结果。
图4显示为本发明实施例2中鼻咽癌队列组织样本肿瘤异质性程度检测结果。
图5显示为本发明实施例2中鼻咽癌队列组织样本肿瘤异质性程度预测免疫治疗疗效结果。
具体实施方式
下面通过具体实施方式结合附图对本发明作进一步详细说明。其中不同实施方式中类似元件采用了相关联的类似的元件标号。在以下的实施方式中,很多细节描述是为了使得本申请能被更好的理解。然而,本领域技术人员可以毫不费力的认识到,其中部分特征在不同情况下是可以省略的,或者可以由其他元件、材料、方法所替代。在某些情况下,本申请相关的一些操作并没有在说明书中显示或者描述,这是为了避免本申请的核心部分被过多的描述所淹没,而对于本领域技术人员而言,详细描述这些相关操作并不是必要的,他们根据说明书中的描述以及本领域的一般技术知识即可完整了解相关操作。
另外,说明书中所描述的特点、操作或者特征可以以任意适当的方式结合形成各种实施方式。同时,方法描述中的各步骤或者动作也可以按照本领域技术人员所能显而易见的方式进行顺序调换或调整。因此,说明书和附图中的各种顺序只是为了清楚描述某一个实施例,并不意味着是必须的顺序,除非另有说明其中某个顺序是必须遵循的。
本文中为部件所编序号本身,例如“第一”、“第二”等,仅用于区分所描述的对象,不具有任何顺序或技术含义。
必须注意,如在本说明书和所附权利要求中所使用的,除非内容另外明确指明,否则单数形式“一种”、“一个”和“该”包含复数指代。
如本文中所用,术语“包括”、“包含”、“含有”及其任何变型旨在涵盖非排他性囊括,以便包括、包含或含有元素或元素列表的过程、方法、方法限定的产品或物质的组合物不仅包含那些元素,而且可以包含这样的过程、方法、方法限定的产品或物质的组合物中没有明确列出的或者并非其固有的其他元素。
如本文中所用,在样本的上下文中使用的术语“提供”意在涵盖获得该样本的任何和全部手段。该术语在实践所要求保护的方法的情况下涵盖导致该样本的存在的所有直接或间接手段。
如本文中所用,术语“患者”优选指人类,但也涵盖其他哺乳动物。术语“生物体”、“个体”、“受试者”、“对象”或“患者”被用作同义词可互换使用。
本发明适用于所有癌症患者。该癌症可以是呼吸系统癌症,或其亚型和分期(phase),呼吸系统包括呼吸道(鼻腔、咽、喉、气管、支气管)和肺,在一些实施方式中,该癌症包括但不限于肺癌、鼻咽癌、喉癌、咽癌,气管癌等。在一些实施方式中,该癌症还可以包括但不限于乳腺癌、肺癌、前列腺癌、结肠直肠癌、脑癌、食道癌、胃癌、膀胱癌、胰腺癌、宫颈癌、头颈癌、卵巢癌、黑素瘤和多药耐药性癌症;或其亚型和分期(phase)。
在一些实施例中,受试者还可以是实体瘤患者,包括但不限于肺癌、鼻咽癌或黑色素瘤患者。
如本文中所用,术语“肿瘤”是指恶性或者良性的所有肿瘤细胞生长和增殖,以及所有的癌前细胞和组织和癌细胞和组织。该癌症包括但不限于呼吸道癌症,该呼吸道癌症包括但不限于肺癌、鼻咽癌、喉癌、咽癌,气管癌等;该癌症还可以是其他淋巴增殖性癌症,例如前体b淋巴母细胞性白血病/淋巴母细胞性淋巴瘤、滤泡性b细胞非霍奇金淋巴瘤、霍奇金淋巴瘤前体t细胞淋巴母细胞性白血病/淋巴母细胞性淋巴瘤、未成熟t细胞的赘生物、外周胸腺后t细胞的赘生物、t细胞幼淋巴细胞白血病、外周t细胞淋巴瘤、未明确的间变性大细胞淋巴瘤、成人t细胞白血病/淋巴瘤、慢性淋巴细胞白血病、套细胞淋巴瘤、滤泡性淋巴瘤、边缘区淋巴瘤、多毛细胞白血病、弥漫性大b细胞淋巴瘤、伯基特淋巴瘤、淋巴浆细胞性淋巴瘤、前体t淋巴母细胞性白血病/淋巴母细胞性淋巴瘤、t细胞幼淋巴细胞性白血病、血管免疫母细胞性淋巴瘤或结节性淋巴细胞为主的霍奇金淋巴瘤。
如本文中所用,变异等位测序深度vi也可称为突变深度,变异等位频率vafi也可称为突变频率。
现有技术或是未能对肿瘤异质性程度进行定量计算,只是定性区分高低程度;或是根据突变频率推算的细胞亚克隆种类的数量进行肿瘤异质性程度的描述,这些估算肿瘤异质性程度的方法都不能够或只能在少量数据集中使得该指标对免疫治疗疗效表现出预测能力。
目前二代测序的成本越来越低,覆盖区域和深度都越来越大,本发明实施例利用全外显子组的大覆盖区域优势,准确检测覆盖区域中的突变位点、突变频率和拷贝数变异情况,利用肿瘤样本中体细胞变异计算肿瘤纯度值,从而计算出肿瘤异质性情况。
在一实施例中,本发明的肿瘤异质性程度评估是在患者用药之前进行的,通过ith这个指标的好坏来评价患者是否能大概率获益于免疫检查点抑制剂anti-pd-(l)1的治疗。由于肺癌的这种免疫治疗总有效率仅为20-25%,患者每年的花费接近20万,若是不能获益,不仅是普通患者家庭经济不能允许,更是社会医疗保险不能承受之重。
在一实施例中,本发明提供了一种预测肿瘤异质性程度的方法,该方法包括以下步骤:s1,通过变异检测软件检测出样本在全外显子范围内的体细胞变异、深度和突变频率信息;s2,通过拷贝数检测软件检测出样本在全外显子范围内的体细胞拷贝数变异,以及估算的肿瘤纯度信息;s3,以s1和s2的计算结果代入到pyclone软件中,根据样本的体细胞变异,以及对应突变位点的测序深度、体细胞变异支持数、拷贝数变异数和肿瘤纯度,将突变按簇进行聚类;s4,根据s3的计算结果,将对应体细胞估算占比最高的突变簇判断为主克隆突变簇,剩余的突变簇判断为亚克隆突变簇;s5,由于pyclone存在体细胞估算占比最高的突变簇仅包含一个突变的异常结果,当发生该情况时,则将对应体细胞估算占比最高和次高的突变簇同时判断为主克隆突变簇,剩余的突变簇判断为亚克隆突变簇;s6,计算肿瘤异质性程度,计算方法为亚克隆突变簇中的体细胞变异数量占所有体细胞变异数量的比值,该值最终的取值范围为[0,1)。
本领域技术人员应当理解,可以通过已知的测序技术对步骤s1中所提及的样本进行测序,包括但不限于全基因组、或探针捕获测序等高通量测序技术及相应的信息学分析方法。
本领域技术人员可以理解,上述实施方式中各种方法的全部或部分功能可以通过硬件的方式实现,也可以通过计算机程序的方式实现。当上述实施方式中全部或部分功能通过计算机程序的方式实现时,该程序可以存储于一计算机可读存储介质中,存储介质可以包括:只读存储器、随机存储器、磁盘、光盘、硬盘等,通过计算机执行该程序以实现上述功能。例如,将程序存储在设备的存储器中,当通过处理器执行存储器中程序,即可实现上述全部或部分功能。另外,当上述实施方式中全部或部分功能通过计算机程序的方式实现时,该程序也可以存储在服务器、另一计算机、磁盘、光盘、闪存盘或移动硬盘等存储介质中,通过下载或复制保存到本地设备的存储器中,或对本地设备的系统进行版本更新,当通过处理器执行存储器中的程序时,即可实现上述实施方式中全部或部分功能。
本发明实施例中,需要同时检测同一受检个体的肿瘤样本和对照样本。受检个体,例如可以是已经通过临床方法确诊为肿瘤患者的个体。肿瘤样本,一般是指来源于肿瘤患者的病患部位或组织的样本,例如肺癌患者的肺部组织样本。对照样本,一般是指来源于同一个肿瘤患者的非病患部位或组织的对照样本,例如外周血分离的白细胞样本等。
本发明实施例中,肿瘤样本和对照样本的基因组二代测序数据一般首先比对到参考基因组上。因此,优选实施例中,在数据获取步骤获取的是肿瘤样本和对照样本的基因组二代测序数据比对到参考基因组的比对文件。在更优选的实施例中,参考基因组具体可以为人参考基因组hg19。
在一些实施例中,如图1所示,本发明提供一种预测肿瘤异质性程度的方法和装置,包括以下方面:
1、高通量测序检测受试者肿瘤采集样本基因变异模块
首先,对选定数个同癌种受试者作为受试对象,对每个受试者进行变异检测和参数计算,具体如下:
1.1通过全基因组、全外显子组或探针捕获测序等高通量测序技术及相应的生物信息学分析方法,对受试者肿瘤组织样本、对照样本进行测序,获取样本中包含的变异,包括但不限于snv、indel、sv、cnv中的至少一种。
在一些实施例中,肿瘤组织样本的测序深度>200×,在另一些实施例中,肿瘤组织样本的测序深度>300×,在另一些实施例中,肿瘤组织样本的测序深度>400×,在另一些实施例中,肿瘤组织样本的测序深度>500×。
在一些实施例中,对照样本的测序深度>50×,在另一些实施例中,对照样本的测序深度>100×,在另一些实施例中,对照样本的测序深度>200×。
1.2根据步骤1.1中的测序结果,获得变异var(变异var选自snv,indel和sv)(vari,i=1,…,n)的参考等位测序深度(ri)和变异等位测序深度(vi),并计算突变位点的变异等位频率(variantallelefraction,vafi),
其中,参考等位测序深度(ri)是测序结果中在相应位点未发生该变异的正常序列的条数,变异等位测序深度(vi)是测序结果中在相应位点发生该变异的变异序列的条数。
在一些实施例中,可以通过现有的变异检测软件获得变异var,所述变异检测软件包括但不限于varscan、mutect,在一些优选的实施例中,所述变异检测软件为varscan(v2.4.1)或mutect(v4.0.12.0)。
1.3利用变异vari所在区域的cnv(cnvi,i=1,…,n)计算变异vari所在区域的参考拷贝数(ncni)和实际总拷贝数(tcni),其中,
同时,通过软件的结果也能获得变异vari所在的两条染色体上等位特异的拷贝数变异(cnvi,major,cnvi,minor,其中cnvi,major≥cnvi,minor,,i=1,…,n),从而计算出实际的等位特异的拷贝数(cni,major,cni,minor),
在一些实施例中,该步骤可以通过现有的检测软件计算变异vari所在区域的参考拷贝数(ncni)和实际总拷贝数(tcni),所述检测软件包括但不限于cnvkit、ascatngs,在一些优选的实施方案中,所述检测软件为cnvkit(v0.8.1)或ascatngs(v3.1.0)。
1.4获得受试者的肿瘤纯度,即肿瘤细胞在采集的肿瘤样本中的占比pur,取值范围为(0,1]。
在一些实施例中,可以通过现有软件获得受试者的肿瘤纯度,所述软件包括但不限于ascatngs,在一些优选的实施方案中,所述软件可以为ascatngs(v3.1.0)软件。
2、肿瘤体细胞变异聚类模块
对每位受试者,依据模块1(即基因变异检测模块)中得到的参数,将检测到的变异进行聚类分析计算。
对于任一类型变异(snv/indel/sv),受试者的采样肿瘤样本中的细胞被分为三类:正常细胞(n)、不携带该变异的肿瘤细胞(twt)和携带该变异的肿瘤细胞(tmut)。携带该变异的肿瘤细胞(tmut))占所有肿瘤细胞(tmut+twt)的比例称为该变异位点的肿瘤细胞比例,如果两个或以上变异位点的变异肿瘤细胞比例相近,那么可以认为它们发生的时间近似,同时很可能发生在相同一群肿瘤细胞中,并且该比例越大,代表在更多的肿瘤细胞中发生该变异,发生的时间也越早。相近比例的变异会被赋予相同的簇标签,聚类成一簇,称为一个克隆。
依次对每个受试者检出的所有个变异(snv/indel/sv)进行聚类,完成该步骤的软件包括但不限于pyclone,优选为pyclone(vo.13.0)。
在一实施例中,使用pyclone(vo.13.0)依次对每个受试者检出的所有个变异(snv/indel/sv)进行聚类,主要参数设置如下:设置--prior参数为major_copy_number,设置--iterations参数为10,000,设置--burn-in参数为1000,设置--tumourcontents参数为受试者肿瘤纯度pur的值,并将受试者的变异结果整理成sample.tsv文件作为参数--in_files的输入。sample.tsv以制表符为分割符的文件,首行为标题行,除标题行外,每行包含一个变异(snv/indel/sv)的信息,包括六列,依次为:变异编号(vari)、参考等位测序深度(ri)、变异等位测序深度(vi)、变异vari所在区域的参考拷贝数(ncni)、变异vari实际的等位特异的拷贝数cni,minor和cni,major这两个参数,其余参数均采用默认参数。
pyclone会根据上述输入信息,估算每个变异vari所在的细胞占所有肿瘤细胞的比例,并依此对每个变异赋予一个簇标签(
在一些实施例中,变异聚类时还可以采用pyclone的其他版本或如clonesig(v0.1)在内的其他变异聚类软件。
3、肿瘤主克隆和亚克隆判断模块
根据上一个模块的计算结果,对每个受试者的每个簇标签(cj,j=1,…,c),都有与之对应的估计肿瘤细胞簇比例
将肿瘤细胞簇比例最高的簇判断为主克隆突变簇(cmain);由于pyclone软件本身算法的特性,有时候会出现肿瘤细胞簇比例最高的簇仅包含一个突变的异常结果,此时则将肿瘤细胞簇比例最高的和次高的簇同时判断为主克隆突变簇cmain,
其中,j=1,...,c,k=1,...,c且k≠j。同时将剩余的簇一同判断为亚克隆突变簇(csub),csub=cl,l∈{1,...,c},l≠j,l≠k。
4、肿瘤异质性程度计算模块
根据上一个模块的判断结果,计算每个受试者的肿瘤异质性程度。统计主克隆突变簇cmain中变异vari的数量nmain和亚克隆突变簇csub中变异vari的数量nsub。肿瘤异质性ith是指亚克隆突变簇中的变异数量占所有变异数量的比值,具体地,
下面将通过具体实施例并结合附图对本发明作进一步说明。应当理解,实施例仅是示例性的,并不构成对本发明保护范围的限制。
实施例1
本实施例中,使用的样本是一批共计69例患者的肺癌免疫治疗队列肿瘤组织样本和对照血液样本,对照血液样本具体为外周血分离的白细胞样本。本实施例的肿瘤组织样本取样方式为对每个患者的肺部病灶处的病理穿刺单点取样并制备的福尔马林固定石蜡包埋(ffpe)样本,取样单位为中山大学肿瘤防治中心。
本实施例成对样本肿瘤异质性检测的具体步骤如下:
1)获取肿瘤组织样本和对照血液样本,进行全外显子组测序,本次测序由南京世和基因生物技术有限公司提供。在得到测序的序列数据后,使用变异检测软件varscan(v2.4.1)检测出受试样本在全外显子范围内的变异(snv/indel),设置最小覆盖率等于20,最小支持读数等于5,其他参数采用默认参数。获取每个突变位点的参考等位测序深度、变异等位测序深度信息。使用拷贝数检测软件ascatngs(v3.1.0)检测出受试样本在全外显子范围内的体细胞拷贝数变异,设置模式为allele_count,其他参数采用默认参数,检测并获得每个等位特异的拷贝数和估算的肿瘤纯度值;
2)根据上一步骤中得到的相关参数,通过pyclone(vo.13.0)软件将突变按簇进行聚类分析,设置--prior参数为major_copy_number,设置--iterations参数为10,000,设置--burn-in参数为1000,设置--tumourcontents参数为受试者肿瘤纯度pur的值。将变异编号、参考等位测序深度、变异等位测序深度、变异所在区域的参考拷贝数、变异实际的等位特异的拷贝数参数制作成制表符为分割符的文件sample.tsv作为--in_files参数的输入,如下举例为这批肺癌队列其中一个患者(患者编号:f17120989277)输入的sample.tsv文件内容。运行pyclone软件后得到队列内各样本的体细胞突变簇的分析结果,如表1所示。
表1
运行pyclone软件后得到队列内各样本的突变聚类的分析结果。如下举例为示例患者f17120989277的pyclone运行结果。其中sample_id为受试者样本编号,cluster_id为pyclone聚类出各变异簇的编号,size为各个变异簇里面包含变异的数量,mean代表估算出的对应变异簇所占肿瘤细胞的比例,std代表计算结果的标准差,具体如表2所示。
表2
3)使用肿瘤主克隆和亚克隆模块,根据上一步体细胞突变簇的分析结果,判断队列内各样本的主克隆突变簇和亚克隆突变簇。
4)使用肿瘤异质性程度计算模块通过上一步结果计算出各样本的肿瘤异质性数值,如图2所示,柱状图代表每个受试患者的肿瘤异质性,纵坐标代表各受试患者的肿瘤异质性数值,红色为亚克隆突变,蓝色为主克隆突变,不同颜色柱子的高低代表克隆占比,亚克隆突变占比越高代表受试肺癌患者的肿瘤异质性越高。
5)在计算出各样本的肿瘤异质性(ith)数值后,取所有肿瘤异质性的中位数,设定ith阈值为0.45,将ith小于等于0.45的样本判断为低ith(ith-l),将ith大于0.45的样本判断为高ith(ith-h)。收集受试患者疗效和无进展生存期(pfs)信息后,如下表3所示,对这批样品进行生存分析(见图3,横坐标的时间单位为天),发现利用ith评估的肿瘤异质性结果,对患者pfs预后有显著的预测效果(p=0.00011),肿瘤异质性高的患者具有更高的进展风险(hr=2.7068)。该结果验证了利用ith分析技术评估肿瘤异质性的有效性和准确性,也说明了ith能够作为生物标志物预测肺癌免疫治疗疗效。
表3
实施例2
本实施例中,使用的样本是一批共计56例患者的鼻咽癌免疫治疗队列肿瘤组织样本和对照血液样本,对照血液样本具体为外周血分离的白细胞样本。本实施例的肿瘤组织样本取样方式为对每个患者的肺部病灶处单点取样。本实施例的肿瘤组织样本取样方式为对每个患者的肺部病灶处的病理穿刺单点取样和制备的福尔马林固定石蜡包埋(ffpe)样本,取样单位为中山大学肿瘤防治中心。
本实施例成对样本肿瘤异质性检测的具体步骤如下:
1)获取肿瘤组织样本和对照血液样本,进行全外显子组测序,本次测序由南京世和基因生物技术有限公司提供。与实施例1相同,在得到测序的序列数据后,使用变异检测软件varscan(v2.4.1)检测出受试样本在全外显子范围内的变异(snv/indel),设置最小覆盖率等于20,最小支持读数等于5,其他参数采用默认参数。获取每个突变位点的参考等位测序深度、变异等位测序深度信息。使用拷贝数检测软件ascatngs(v3.1.0)检测出受试样本在全外显子范围内的体细胞拷贝数变异,设置模式为allele_count,其他参数采用默认参数,检测并获得每个等位特异的拷贝数和估算的肿瘤纯度信息。
2)根据上一步骤中得到的相关参数,通过pyclone(vo.13.0)软件将突变按簇进行聚类分析,设置--prior参数为major_copy_number,设置--iterations参数为10,000,设置--burn-in参数为1000,设置--tumourcontents参数为受试者肿瘤纯度pur的值。将变异编号、参考等位测序深度、变异等位测序深度、变异所在区域的参考拷贝数、变异实际的等位特异的拷贝数参数制作成制表符为分割符的文件sample.tsv作为--in_files参数的输入,如下举例为这批鼻咽癌队列其中一个受试患者(患者编号:f17120989297)输入的sample.tsv文件内容。运行pyclone软件后得到队列内各样本的体细胞突变簇的分析结果,如表4所示。
表4
运行pyclone软件后得到队列内各样本的突变聚类的分析结果。如下举例为示例患者的pyclone运行结果。其中sample_id为受试者样本编号,cluster_id为pyclone聚类出各变异簇的编号,size为各个变异簇里面包含变异的数量,mean代表估算出的对应变异簇所占肿瘤细胞的比例,std代表计算结果的标准差,该样本的突变聚类的分析结果如表5所示。
表5
3)使用肿瘤主克隆和亚克隆模块,根据上一步体细胞突变簇的分析结果,判断队列内各样本的主克隆突变簇和亚克隆突变簇。
4)使用肿瘤异质性程度计算模块通过上一步结果计算出各样本的肿瘤异质性数值,如图4所示,柱状图代表每个受试患者的肿瘤异质性,红色为亚克隆突变,蓝色为主克隆突变,不同颜色柱子的高低代表克隆占比,亚克隆突变占比越高代表受试鼻咽癌患者的肿瘤异质性越高。
5)在计算出各样本的肿瘤异质性(ith)数值后,取所有肿瘤异质性的中位数,经过roc曲线分析校正后,设定ith阈值为0.33,将ith小于等于0.33的样本判断为低ith(ith-l),将ith大于0.33的样本判断为高ith(ith-h)。收集受试患者疗效和无进展生存期(pfs)信息后,如下表6所示,对这批样品进行生存分析(见图5,横坐标的时间单位为天),发现利用ith评估的肿瘤异质性结果,对患者pfs预后有显著的预测效果(p=0.016),肿瘤异质性高的患者具有更高的进展风险(hr=2.0501)。该结果重新在另一个鼻咽癌队列中成功验证了利用ith分析技术评估肿瘤异质性的有效性和准确性,也说明了ith能够作为生物标志物在鼻咽癌中预测免疫治疗的疗效。
表6
综上,本发明的实施例基于全外显子组测序(wes)的数据提供了一种计算样本的肿瘤异质性程度的具体方法,充分利用样本各突变位点检测的突变频率估算出对应肿瘤克隆的细胞占比,从而推算出肿瘤异质性程度的具体数值,为后续预测免疫治疗疗效提供了一个数值上可以参考的依据。
以上应用了具体个例对本发明进行阐述,只是用于帮助理解本发明,并不用以限制本发明。对于本发明所属技术领域的技术人员,依据本发明的思想,还可以做出若干简单推演、变形或替换。
- 还没有人留言评论。精彩留言会获得点赞!