小檗碱类化合物在制备抗肿瘤药物中的应用
1.本发明涉及药物化学技术领域,具体涉及一种小檗碱类化合物在制备抗肿瘤药物中的应用。
背景技术:
2.小檗碱,一种以异喹啉为母核的天然生物碱,具有抑制脲酶活性、抗炎、抗氧化剂、抗阿尔茨海默效应和抑制细胞色素p450酶活性等广泛的药理作用。
3.组蛋白赖氨酸特异性去甲基化酶1(lsd1)是第一个被发现的组蛋白赖氨酸去甲基酶,与包括细胞增殖和分化、胚胎多能性和癌症发生发展等密切相关。研究发现lsd1在许多肿瘤细胞中过表达,从而导致各种癌症的发生,如白血病、肺癌等。lsd1抑制剂通过抑制肿瘤细胞中lsd1的活性,进而影响肿瘤细胞的生长、转移和侵袭,因此设计并合成高选择性、高效、低毒的lsd1抑制剂是治疗癌症新途径。
4.过去的十几年,lsd1抑制剂的开发取得了重大进展。目前,已有多种用于治疗小细胞肺癌(sclc)和急性髓细胞性白血病(aml)的苯环丙胺(tcp)类不可逆抑制剂正在进行临床实验,包括tcp,gsk2879552,img
‑
7289,ory
‑
1001,incb059872和ory
‑
2001。而可逆抑制剂中,目前仅有cc
‑
90011和sp
‑
2577进入临床研究阶段,因此发现具有不同化学结构和显著生物活性的新型可逆lsd1抑制剂已成为癌症治疗的一种很有前途的策略。
5.但是将小檗碱类化合物用于以lsd1为靶点的抗肿瘤研究的报道较少,因此此类研究具有重要的意义。
技术实现要素:
6.有鉴于此,为充分开发利用现有的天然产物资源,本发明提供一种小檗碱类化合物的新用途,以解决上述问题。
7.本发明的一个目的在于提供一种小檗碱类化合物在制备抗肿瘤药物中的应用,为寻找基于lsd1靶点的抗肿瘤药物开辟一条新途径。
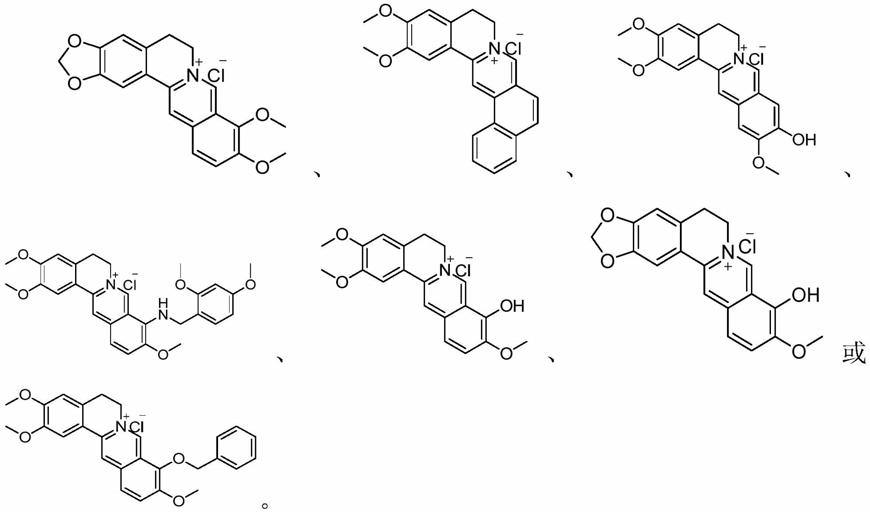
8.具体地,一种小檗碱类化合物在制备抗肿瘤药物中的应用,其中,所述抗肿瘤药物为lsd1抑制剂,所述小檗碱类化合物为盐酸小檗碱类化合物、四氢小檗碱类化合物或二氢小檗碱类化合物。
9.基于上述,所述盐酸檗碱类化合物的结构通式可以为优选地,所述盐酸檗碱类化合物的结构式为:
10.基于上述,所述四氢小檗碱类化合物的结构通式可以为优选地,所述四氢小檗碱类化合物的结构式为:
[0011][0012]
基于上述,所述二氢小檗碱类化合物的结构式为:
[0013]
本发明提供的一种小檗碱类化合物在制备抗肿瘤药物中的应用,其中的小檗碱类化合物是一类以异喹啉为母核的衍生物,经验证,其具有抑制lsd1的活性,能够用作制备lsd1抑制剂,为寻找基于lsd1靶点的抗肿瘤药物开辟一条新途径。
具体实施方式
[0014]
下面通过具体实施方式,对本发明的技术方案做进一步的详细描述。若未特别指明,以下实施例中所用的技术手段为本领域技术人员所熟知的常规手段。
[0015]
实施例一
[0016]
本实施例提供化合物1,该化合物1的结构式为:
[0017]
该化合物1的制备路线如下:
[0018][0019]
该化合物1的制备方法包括以下步骤:
[0020]
(1)化合物1
‑
3的制备:
[0021]
将2,3
‑
二甲氧基苯甲醛(化合物1
‑
1)0.50g(约3.01mmol)和2
‑
[3,4
‑
(亚甲基二氧)苯基]乙胺(化合物1
‑
2)0.50g(约3.01mmol)加入10ml甲醇中,常温反应2h,缓慢加入硼氢化钠0.17g(约4.51mmol),并于室温下反应,采用tlc监测该反应体系。待反应完全后,用乙酸乙酯和饱和碳酸钾水溶液萃取,合并有机相,无水硫酸镁干燥,蒸干,经柱层析得化合物1
‑
3,该化合物1
‑
3为淡黄色液体,收率约为81%。
[0022]
(2)化合物1的制备:
[0023]
将0.5g(约1.59mmol)上述化合物1
‑
3加入反应瓶,加入甲酸溶液5ml,加入无水硫酸铜1.01g(约6.34mmol),50%乙二醛水溶液0.37mg(约3.17mmol)并于80℃下加热回流,采用tlc监测该反应体系。反应结束后立即抽滤,滤液加入乙酸乙酯,即有大量黄色固体析出,抽滤得化合物1
‑
4,为黄色固体。
[0024]
将上述该黄色固体加入至10ml甲醇中,加入0.5ml浓盐酸酸化,常温搅拌12h,蒸干,炒样,经柱层析得化合物1。该化合物为黄色固体,该化合物1的收率为53%。1hnmr(400mhz,dmso
‑
d6)δ9.90(s,1h),8.96(s,1h),8.20(d,j=8.9hz,1h),8.01(d,j=8.9hz,1h),7.79(s,1h),7.09(s,1h),6.17(s,2h),4.94(t,j=6.2hz,2h),4.10(s,3h),4.07(s,3h),3.21(t,j=6.2hz,2h)。
[0025]
实施例二
[0026]
本实施例提供化合物2,该化合物2的结构式为
[0027]
该化合物2制备路线如下:
[0028][0029]
该化合物2的制备方法包括以下步骤:
[0030]
(1)化合物2
‑
3的制备:
[0031]
将2
‑
萘甲醛(化合物2
‑
1)0.50g(约3.20mmol)和3,4
‑
二甲氧基苯乙胺(化合物2
‑
2)0.58g(约3.20mmol)加入10ml甲醇中,常温反应2h,缓慢加入硼氢化钠0.18g(约4.80mmol),并于室温下反应,采用tlc监测该反应体系。待反应完全后,用乙酸乙酯和饱和碳酸钾水溶液萃取,合并有机相,无水硫酸镁干燥,蒸干,经柱层析得化合物2
‑
3,该化合物2
‑
3为淡黄色液体,收率约为89%。
[0032]
(2)化合物2的制备:
[0033]
将0.5g(约1.56mmol)上述化合物2
‑
3加入反应瓶,加入甲酸溶液5ml,加入无水硫酸铜0.99g(约6.22mmol),50%乙二醛水溶液0.36mg(约3.11mmol)并于80℃下加热回流,采用tlc监测该反应体系。反应结束后立即抽滤,滤液加入乙酸乙酯,即有大量黄色固体析出,抽滤得化合物2
‑
4,为黄色固体。
[0034]
将上述该黄色固体加入至10ml甲醇中,加入0.5ml浓盐酸酸化,常温搅拌12h,蒸干,炒样,经柱层析得化合物2。该化合物为黄色固体,该化合物2的收率为65%。1hnmr(400mhz,dmso
‑
d6)δ9.92(s,1h),9.55(s,1h),9.41(d,j=7.8hz,1h),8.27
–
8.19(m,2h),8.13(d,j=9.0hz,1h),8.06
–
7.95(m,3h),7.17(s,1h),4.92(t,j=6.5hz,2h),4.04(s,3h),3.91(s,3h),3.30(t,j=6.5hz,2h)。
[0035]
实施例三
[0036]
本实施例提供化合物3,该化合物3的结构式为
[0037]
该化合物3的制备路线如下:
[0038][0039]
该化合物3的制备方法包括以下步骤:
[0040]
(1)化合物3
‑
1的制备:
[0041]
将1.00g 3
‑
羟基
‑4‑
甲氧基苯甲醛(约6.57mmol)和1.36g(约9.86mmol)的碳酸钾分别加入到反应瓶中,然后加入约10ml n,n
‑
二甲基甲酰胺,将1.69g(约9.86mmol)溴化苄加入以上体系,并在80℃加热回流下,采用薄层层析tlc监测反应。反应结束后,用乙酸乙酯和水萃取,合并有机相,蒸干,炒样,经柱层析得到纯品化合物3
‑
1。
[0042]
(2)化合物3
‑
3的制备:
[0043]
将0.50g(约2.06mmol)化合物3
‑
1和3,4
‑
二甲氧基苯乙胺(化合物1
‑
2)0.38g(约0.26mmol)加入10ml甲醇中,常温反应2h,缓慢加入硼氢化钠0.12g(约3.10mmol),并于室温下反应,采用tlc监测该反应体系。待反应完全后,用乙酸乙酯和饱和碳酸钾水溶液萃取,合并有机相,无水硫酸镁干燥,蒸干,经柱层析得化合物3
‑
3,该化合物3
‑
3为淡黄色液体,收率约为89%。
[0044]
(3)化合物3的制备:
[0045]
将0.5g(约1.32mmol)上述化合物3
‑
3加入反应瓶,加入甲酸溶液5ml,加入无水硫酸铜0.85g(约5.30mmol),50%乙二醛水溶液0.31mg(约2.65mmol)并于80℃下加热回流,采用tlc监测该反应体系。反应结束后立即抽滤,滤液加入乙酸乙酯,即有大量黄色固体析出,抽滤得化合物3
‑
4,为黄色固体。
[0046]
将上述该黄色固体加入至10ml甲醇中,加入0.5ml浓盐酸酸化,常温搅拌12h,蒸干,炒样,经柱层析得化合物3。该化合物3为黄色固体,该化合物3的收率为24%。1hnmr(400mhz,dmso
‑
d6)δ9.55(s,1h),8.81(s,1h),7.66(s,1h),7.61(s,1h),7.53(s,1h),7.09
(s,1h),4.75(t,j=6.2hz,2h),4.08(s,3h),3.93(s,3h),3.87(s,3h),3.21(t,j=6.2hz,2h)。
[0047]
实施例四
[0048]
本实施例提供化合物4,该化合物4的结构式为
[0049]
该化合物4的制备路线如下:
[0050][0051]
该化合物4的制备方法包括以下步骤:
[0052]
(1)化合物4
‑
3的制备:
[0053]
将2,3
‑
二甲氧基苯甲醛(化合物1
‑
1)0.46g(约2.76mmol)和3,4
‑
二甲氧基苯乙胺(化合物1
‑
2)0.5g(约2.76mmol)加入10ml甲醇中,常温反应2h,缓慢加入硼氢化钠0.16g(约4.14mmol),并于室温下反应,采用tlc监测该反应体系。待反应完全后,用乙酸乙酯和饱和的碳酸钾水溶液萃取,合并有机相,无水硫酸镁干燥,蒸干,经柱层析得化合物4
‑
3,该化合物4
‑
3为淡黄色液体,收率约为92%。
[0054]
(2)化合物4
‑
4和4
‑
5的制备:
[0055]
将0.5g(约1.51mmol)上述化合物4
‑
3加入反应瓶,加入甲酸溶液5ml,加入无水硫酸铜0.97g(约6.03mmol),50%乙二醛水溶液0.35ml(约3.02mmol)并于80℃下加热回流,采用tlc监测该反应体系。反应结束后立即抽滤,滤液加入乙酸乙酯,即有大量黄色固体析出,抽滤得化合物4
‑
4,为黄色固体。将上述该黄色固体加入至10ml甲醇中,加入0.5ml浓盐酸酸化,常温搅拌12h,炒样,经柱层析得化合物4
‑
5。
[0056]
(3)化合物4的制备:
[0057]
将0.5g化合物4
‑
4(约1.29mmol),3,4
‑
二甲氧基苄胺(15
‑
a)7.5ml(约49.7mmol)加入反应瓶中,于100℃下回流4h,采用tlc监测该反应体系。待反应完全后,用乙酸乙酯洗去过量的3,4
‑
二甲氧基苄胺,得红色固体,炒样,经柱色谱得化合物4。该化合物4为深红色固体,且经检测计算其收率约为65%,1hnmr(400mhz,methanol
‑
d4)δ9.60(s,1h),8.64(s,1h),7.87(d,j=8.8hz,1h),7.69(d,j=8.8hz,1h),7.64(s,1h),7.11(d,j=8.3hz,1h),7.05(s,1h),6.53(d,j=2.4hz,1h),6.42(dd,j=8.3,2.4hz,1h),4.81(t,j=6.3hz,2h),4.65(s,2h),4.00(s,3h),3.98(s,3h),3.95(s,3h),3.79(s,3h),3.76(s,3h),3.29(t,j=
6.3hz,2h)。
[0058]
实施例五
[0059]
本实施例提供化合物5,该化合物5的结构式为
[0060]
该化合物5的制备路线如下:
[0061][0062]
该化合物5的制备方法包括以下步骤:
[0063]
(1)化合物4
‑
3、4
‑
4、4
‑
5的制备同实施例四。
[0064]
(2)化合物5的制备:
[0065]
将0.5g化合物4
‑
4在真空条件(30~40mmhg)下,于190℃下加热30min得红黑色固体,炒样,柱色谱得化合物5。化合物5为深红色固体,且经检测计算其收率约为76%,1hnmr(400mhz,dmso
‑
d6)δ9.18(s,1h),8.15(s,1h),7.53(s,1h),7.30(d,j=7.9hz,1h),6.98(s,1h),6.51(d,j=7.9hz,1h),4.54(t,j=6.0hz,3h),3.89(s,3h),3.83(s,3h),3.76(s,3h),3.07(t,j=6.0hz,3h)。
[0066]
实施例六
[0067]
本实施例提供化合物6,该化合物6的结构式为
[0068]
该化合物6的制备路线如下:
[0069][0070]
该化合物6的制备方法包括以下步骤:
[0071]
(1)化合物1
‑
3、1
‑
4和1的制备同实施例一。
[0072]
(2)化合物6的制备:
[0073]
在真空条件(30~40mmhg)下,将0.5g化合物1于190℃下加热30min得红黑色固体,炒样,柱色谱得化合物6。化合物6为深红色固体,且经检测计算其收率约为85%,1hnmr(400mhz,dmso
‑
d6)δ9.07(s,1h),7.98(s,1h),7.61(s,1h),7.21(d,j=7.9hz,1h),6.96(s,1h),6.35(d,j=7.8hz,1h),6.10(s,2h),4.48(t,j=6.1hz,2h),3.73(s,3h),3.04(t,j=6.1hz,2h)。
[0074]
实施例七
[0075]
本实施例提供化合物7,该化合物7的结构式为
[0076]
该化合物7的结制备路线如下:
[0077][0078]
该化合物7的制备方法包括以下步骤:
[0079]
(1)化合物4
‑
3、4
‑
4和4
‑
5的制备同实施例四。
[0080]
(2)化合物5的制备同实施案五。
[0081]
(3)化合物7的制备:
[0082]
将0.2g化合物5(约0.54mmol)和0.08g(约0.82mmol)的三乙胺分别加入到反应瓶中,然后加入约10ml二氯甲烷,将1.69g(约0.54mmol)溴化苄加入以上体系,并常温搅拌,采用薄层层析tlc监测反应。反应结束后,过滤,所得滤饼炒样,经柱层析得到纯品化合物5。产率为90%,1hnmr(400mhz,dmso
‑
d6)δ9.74(s,1h),9.05(s,1h),8.23(d,j=9.2hz,1h),8.05(d,j=9.2hz,1h),7.72(s,1h),7.61
–
7.58(m,2h),7.44
–
7.35(m,3h),7.10(s,1h),5.37(s,2h),4.94(t,j=6.3hz,2h),4.10(s,3h),3.94(s,3h),3.87(s,3h),3.22(t,j=6.3hz,2h)。
[0083]
实施例八
[0084]
本实施例提供化合物8,该化合物8的结构式为
[0085]
该化合物8的制备路线如下:
[0086][0087]
该化合物8的制备方法包括以下步骤:
[0088]
(1)化合物4
‑
3、4
‑
4和4
‑
5的制备同实施例四。
[0089]
(2)化合物8的制备:
[0090]
将0.2g(约0.54mmol)化合物4
‑
4加入10ml甲醇溶液中,加热至45℃,加入nabh
4 0.081g(约2.14mmol),采用tlc监测该反应体系。待反应完全后,用乙酸乙酯和饱和碳酸钾水溶液萃取,合并有机相,无水硫酸镁干燥,蒸干,经柱层析得化合物8。该化合物为白色固体,收率约为81%。1hnmr(400mhz,dmso
‑
d6)δ8.58(s,1h),6.87(s,1h),6.79(d,j=8.3hz,1h),6.68(s,1h),6.60(d,j=8.3hz,1h),4.02(d,j=15.7hz,1h),3.76(s,3h),3.74(s,3h),3.72(s,3h),3.42
–
3.35(m,2h),3.29(d,j=15.7hz,2h),3.12
–
3.05(m,1h),2.99
–
2.86(m,1h),2.64
–
2.52(m,2h),2.49
–
2.39(m,1h).
[0091]
实施例九
[0092]
本实施例提供化合物9,该化合物9的结构式为
[0093]
该化合物9的制备路线如下:
[0094][0095]
该化合物9的制备方法包括以下步骤:
[0096]
(1)化合物1
‑
3,1
‑
4,1的制备同实施例一
[0097]
(2)化合物9的制备:
[0098]
将0.2g(约0.54mmol)化合物1加入10ml甲醇溶液中,加热至45℃,加入nabh
4 0.081g(约2.15mmol),采用tlc监测该反应体系。待反应完全后,用乙酸乙酯和饱和碳酸钾水溶液萃取,合并有机相,无水硫酸镁干燥,蒸干,经柱层析得化合物9。该化合物为白色固体,收率约为86%。1hnmr(400mhz,dmso
‑
d6)δ8.58(s,1h),6.91(s,1h),6.78(d,j=8.2hz,1h),6.66(s,1h),6.57(d,j=8.2hz,1h),5.94(d,j=2.6hz,2h),4.01(d,j=15.6hz,1h),3.76(s,3h),3.43
–
3.35(m,2h),3.27(d,j=15.6hz,2h),3.12
–
3.03(m,1h),2.95
–
2.85(m,1h),2.63
–
2.51(m,2h),2.48
–
2.40(m,1h)。
[0099]
实施例十
[0100]
本实施例提供化合物10,该化合物10的结构式为
[0101]
该化合物10的制备路线如下:
[0102][0103]
该化合物10的制备方法包括以下步骤:
[0104]
(1)化合物10
‑
1的制备
[0105]
将0.50g 3,4
‑
二羟基苯甲醛(约3.62mmol),将0.75g二溴甲烷(约4.34mmol),1.37g(约4.93mmol)的碳酸钾分别加入到反应瓶中,然后加入约10ml n,n
‑
二甲基甲酰胺,并在90℃加热回流下,采用薄层层析tlc监测反应。反应结束后,用乙酸乙酯和水萃取,合并有机相,蒸干,炒样,经柱层析得到纯品化合物10
‑
1。
[0106]
(2)化合物10
‑
3和10
‑
4的制备
[0107]
将0.5g(约1.59mmol)上述化合物10
‑
3加入反应瓶,加入甲酸溶液5ml加入无水硫酸铜1.01g(约6.34mmol),50%乙二醛水溶液0.37mg(3.17mmol)并于80℃下加热回流,采用tlc监测该反应体系。反应结束后立即抽滤,滤液加入乙酸乙酯,即有大量黄色固体析出,抽滤得化合物10
‑
4,化合物10
‑
4为黄色固体。将上述该黄色固体加入至10ml甲醇中,加入0.5ml浓盐酸酸化,常温搅拌12h,蒸干,炒样,经柱层析得化合物10
‑
5。
[0108]
(3)化合物10的制备
[0109]
将0.2g(约0.54mmol)化合物10
‑
4加入10ml甲醇溶液中,加热至45℃,加入nabh
4 0.081g(约2.15mmol),采用tlc监测该反应体系。待反应完全后,用乙酸乙酯和饱和碳酸钾水溶液萃取,合并有机相,无水硫酸镁干燥,蒸干,经柱层析得化合物10,该化合物为白色固体,收率为79%。1hnmr(400mhz,chloroform
‑
d)δ6.73(s,1h),6.71
–
6.67(m,1h),6.67
–
6.64(m,1h),6.62(s,1h),5.97(d,j=1.5hz,1h),5.93(d,j=1.5hz,1h),4.11(d,j=15.3hz,1h),3.89(s,3h),3.87(s,3h),3.64
–
3.49(m,2h),3.28(dd,j=16.0,3.7hz,1h),3.21
–
3.08(m,2h),2.82(dd,j=15.9,11.3hz,1h),2.72
–
2.60(m,2h)。
[0110]
实施例十一
[0111]
本实施例提供化合物11,该化合物11的结构式为
[0112]
该化合物11的制备路线如下:
[0113][0114]
该化合物11的制备方法包括以下步骤:
[0115]
(1)化合物10
‑
1、10
‑
3和10
‑
4的制备步骤同实施例10。
[0116]
(2)化合物11的制备:
[0117]
将0.2g(约0.54mmol)化合物10
‑
4和0.22g k2co3(约1.61mmol)加入5ml甲醇溶液中,并将含0.03mgnabh4(0.16mmol)的5%naoh水溶液滴加至以上体系,采用tlc监测该反应体系。待反应完全后,用乙酸乙酯和水溶液萃取,合并有机相,无水硫酸镁干燥,蒸干,经柱层析得化合物11,该化合物为黄色固体,产率为65%。1hnmr(400mhz,dmso
‑
d6)δ7.19(s,1h),6.98(d,j=8.4hz,1h),6.88(s,j=8.4hz,1h),6.65(s,1h),5.76(s,1h),4.11(s,2h),3.89(s,3h),3.87(s,3h),3.64
–
3.49(m,2h),3.21
–
3.08(m,2h)。
[0118]
lsd1抑制活性测定
[0119]
实验方法:样品为上述实施例合成的化合物1~11和ory
‑
1001(购买自medchemexpress公司)。样品储备液:分别称取样品1~2mg,用dmso(二甲基亚砜)溶解成浓度为10mm的母液,实验时用dmso稀释至需要测定浓度。分别将样品与从大肠杆菌表达体系中纯化获得的人源复合体蛋白lsd1/corest孵育,随后加入由吉尔生化(上海)有限公司合成的底物h3k4me2孵育反应30min。孵育结束后,加入荧光染料amplex red和辣根过氧化物酶hrp反应5min,随后使用envision酶标仪(perkinelmer,waltham,ma,usa)测定荧光信号(e
x
=535nm,e
m
=595nm),并计算其抑制率,结果如表1所示。具体地,抑制率计算公式如下:
[0120][0121]
其中,上述抑制率计算公式中的“样品组荧光强度”、“标准组荧光强度”和“空白组荧光强度”均是采用参照上述实验方法测得的荧光强度数值,不同之处在于:在相同条件下,“样品组荧光强度”的测定对象是含待测上述化合物的样品液,“标准组荧光强度”的测定对象是标准液,且该标准液与所述样品液相比不含有样品。“空白组荧光强度”的测定对象是不含lsd1/corest复合体蛋白和h3k4me2多肽的空白样品。用graphpad prism8.0处理上述样品化合物的ic
50
数据,结果如表1所示。
[0122]
表1本发明实施例提供的化合物对lsd/corest的抑制活性结果表
[0123][0124]
最后应当说明的是:以上实施例仅用以说明本发明的技术方案而非对其限制;尽管参照较佳实施例对本发明进行了详细的说明,所属领域的普通技术人员应当理解:依然可以对本发明的具体实施方式进行修改或者对部分技术特征进行等同替换;而不脱离本发明技术方案的精神,其均应涵盖在本发明请求保护的技术方案范围当中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1