环糊精‑甲基乙烯基醚/马来酸酐共聚物及其自组装纳米粒在口服药物传递中应用的制造方法与工艺
环糊精-甲基乙烯基醚/马来酸酐共聚物及其自组装纳米粒在口服药物传递中应用技术领域本发明属于药物制剂新辅料和新剂型领域,涉及多功能聚酸酐类载体材料环糊精-甲基乙烯基醚/马来酸酐共聚物及其在难溶性药物的口服传递中的应用。
背景技术:
迄今为止,口服给药仍然是人们的首选方式,这得益于其良好的患者顺应性。然而,许多候选药物,由于胃酸的破坏、黏液层截留和渗透、外排蛋白对药物渗透性的调控以及系统前代谢(肠道和肝脏首过代谢)等因素的存在,难以获得理想的口服吸收,无法实现临床化。鉴于口服传递的复杂性,往往需要多重策略的联合应用以实现明显提高药物的口服生物利用度。环糊精(CD)是由环糊精糖苷酶作用于淀粉,生成的环状多糖化合物。环糊精的基本结构是上窄下宽两端开口的环状中空筒形分子,它具有亲水性的外表面和疏水的内腔。传统的环糊精的研究主要集中在对环糊精-药物包合物的研究上,环糊精主要作为疏水性药物的增溶剂,用于增加药物的稳定性和生物利用度、降低毒副作用等。近年来,研究表明环糊精还具有抑制P-糖蛋白(P-gp)的作用,该作用的主要被认为是由环糊精分子与细胞膜上胆固醇分子的相互作用引起的。此外,研究表明环糊精还具有抑制多种CYP450酶的作用。然而,环糊精在胃肠道中透膜吸收能力不佳,对难溶性药物增溶能力亦有限。聚酸酐类高分子聚合物在体内具有良好的生物相容性。甲基乙烯基醚/马来酸酐共聚物(PVM/MA)作为该类高分子聚合物中的一种,生物粘附性是其显著的特点。另外,分子内酸酐键作为准羧酸基团,便于对其进行接枝修饰,实现功能化的补充,获得多功能性载体材料。本发明结合上述两种辅料的特点,通过酯化反应,首次合成了两亲性的环糊精-甲基乙烯基醚/马来酸酐共聚物(CD-PVM/MA),并将其自组装形成的纳米粒应用于难溶性药物的口服吸收传递。
技术实现要素:
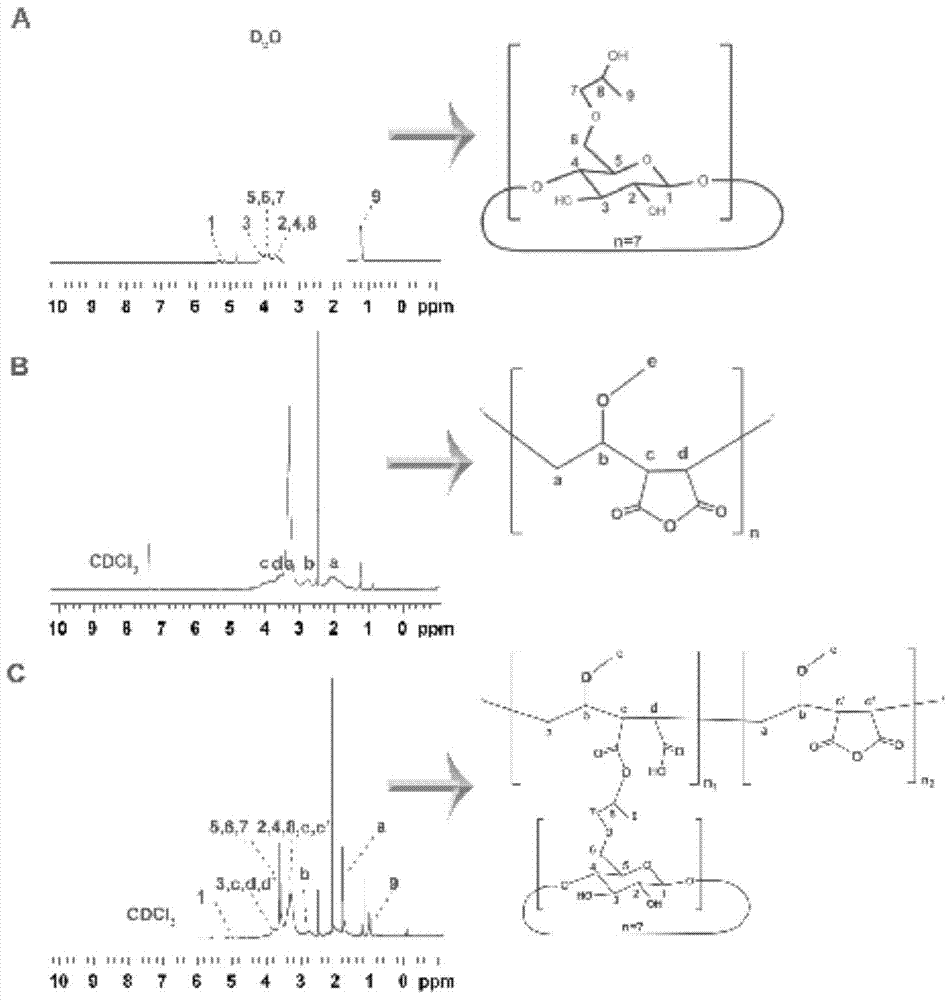
本发明旨在提供一种两亲性的多功能聚酸酐类载体材料环糊精-甲基乙烯基醚/马来酸酐共聚物(CD-PVM/MA),生物粘附性和P-gp与CYP450双重抑制功能为其显著特征。本发明第二个目的旨在提供CD-PVM/MA的合成方法。本发明的第三个目的旨在提供CD-PVM/MA自组装形成的纳米粒在药物传递中的应用。本发明通过以下技术方案实现上述目的:CD-PVM/MA是以水溶性环糊精及其衍生物为亲水端,以酯键链接到疏水性的聚酸酐主链上而形成两亲性的载体材料,其中PVM/MA的生物粘附性得以保留,另外,引入了环糊精对P-gp及CYP450的抑制功能。所述的CD-PVM/MA的结构式通式如下:所述的CD-PVM/MA,其特征在于所用的PVM/MA的分子量范围10万-250万,优选10万-100万,更优选10万-50万,最优选10万-20万,环糊精可为α,β,γ环糊精及其衍生物,优选β-环糊精,所述的β-环糊精选自羟丙基-β-环糊精,氨基-β-环糊精。n2=35n1,n1+n2=n/156,n为PVM/MA的分子量。其制备过程:将PVM/MA和1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC)溶于有机溶剂中,冰浴活化后,加入环糊精,于室温(25℃)和氮气保护的条件下进行反应,经分离纯化后得到白色粉末,即为CD-PVM/MA。反应式如下:所述的CD-PVM/MA兼具PVM/MA和环糊精的特征,实现了多功能的一体化。所述的聚合物胶束可采用超声法或乳化溶剂挥发法制备,其特征在于采用下述步骤:将CD-PVM/MA完全溶解于少量的丙酮/四氢呋喃混合溶剂中,缓慢滴加至含有乙醇的蒸馏水中,探头超声后,旋转蒸发除去有机溶剂,即得空白纳米粒溶液。含药纳米粒溶液的制备,首先将疏水性药物和CD-PVM/MA溶解于混合溶剂中,其余操作同空白纳米粒的制备。所述的疏水性药物选自:他克莫司,紫杉醇,阿霉素,羟基喜树碱,多西他赛等。本发明具有以下有益效果:本发明提供了一种新的药物载体CD-PVM/MA,其制备过程温和,易操作。其可进一步制备成纳米粒制剂,制备简便,粒径较小且均一,稳定性好,可作为多种难溶性药物的载体,与药物组合后形成一种药物传递系统,该系统兼具生物粘附性和外排蛋白(P-gp)与代谢酶(CYP450)双重抑制作用,并可促进药物的淋巴转运,降低肝脏的首过效应,最终实现生物利用度的显著提高。本发明的载体尤其适用于提高本身为P-gp和/或CYP450底物类难溶性药物的口服生物利用度。附图说明图1为本发明实施例1中CD-PVM/MA的合成过程示意图。图2为本发明实施例1中CD-PVM/MA的1HNMR谱图。图3为本发明实施例1中CD-PVM/MA的IR谱图。图4为本发明实施例4中他克莫司聚酸酐纳米粒的示意图、外观图、透视电镜图及动态光散射测定纳米粒粒径图。图5为本发明实施例5中聚酸酐纳米粒肠道分布图。图6为本发明实施例6中他克莫司聚酸酐纳米粒抑制P-gp实验结果图。图7为本发明实施例7中他克莫司聚酸酐纳米粒淋巴转运实验结果图。图8为本发明实施例8中他克莫司聚酸酐纳米粒口服生物利用度结果图。图9为本发明实施例9中他克莫司聚酸酐纳米粒口服生物利用度结果图(PVM/MA,分子量:10万)。具体实施方式下面通过实施例的方式进一步说明本发明,但并不因此将发明限制在所述的实施例范围之中。实施例1以羟丙基β环糊精(HP-β-CD)和PVM/MA(分子量:20万)为原料合成CD-PVM/MA。将PVM/MA和1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC)溶于丙酮/四氢呋喃(3/2,v/v)中,冰浴活化2小时后,加入HP-β-CD(摩尔量为PVM/MA的36倍),于室温(25℃)和氮气保护的条件下进行反应3小时,经分离纯化后得到白色粉末,即为CD-PVM/MA。反应式如下:实施例2反应式中的PVM/MA的分子量范围10万-250万,环糊精可为α,β,γ环糊精及其衍生物。采用核磁共振测定1HNMR氢谱来确定实施例1中CD-PVM/MA的结构,PVM/MA和CD-PVM/MA以氘带二氯甲烷为溶剂,HP-β-CD以重水为溶剂,结果见图2。CD的信号峰归属为3.5-4.0ppm(羟基葡萄糖环C3、4、5、6位次甲基);PVM/MA信号峰归属如下:3.567ppm(母链上次甲基),2.513ppm(乙酯基),2.181ppm(环酸酐上的氢)。与PVM/MA的核磁共振图谱比较,环糊精接枝的PVM/MA核磁图谱3.5-4.0ppm出现裂分峰(羟基环糊精信号峰),而且核磁图谱向高场位移,表明环糊精已接枝到PVM/MA上。实施例3采用红外图谱来确定实施例1中CD-PVM/MA的结构,选用KBr为空白辅料,结果如图3。比较CD...
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1