产生人抗体的细胞的制作方法
本发明涉及产生人抗体的细胞。更具体而言,涉及产生人抗体的禽B细胞。
背景技术:
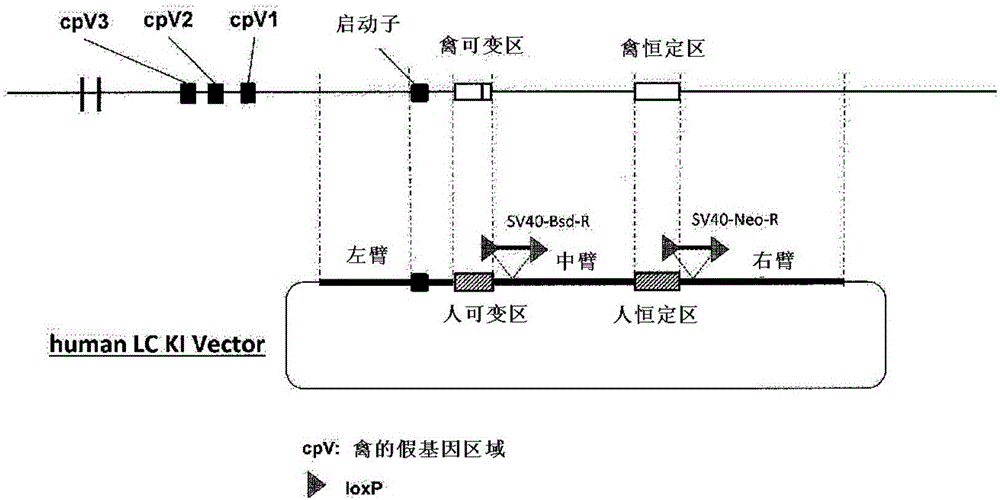
:抗体在进行生物物质的确定、功能解析等方面是有用的,此外,在疾病的治疗中抗体发挥的作用也大。抗体在生物体内与特定的抗原结合,引起各种生物体内防御反应,例如抗体依赖性细胞伤害(ADCC)活性、补体依赖性细胞伤害(CDC)活性,识别对于生物体而言的“异物”,进行除去。着眼于这样的抗体的能力,抗体医药的开发近年来已经积极地进行。在进行多种多样的疾病的治疗时,需要迅速、简便提供识别各种靶抗原的抗体。目前为止,作为制备具有期望的物理学、生理学特征的抗体组的方法,已使用单克隆抗体的制造技术。制造单克隆抗体时,通常采用使利用生物体内免疫产生的B细胞与骨髓瘤融合的杂交瘤法。但是,该方法除了由于生物体内免疫的利用而产生的免疫耐受之外,还存在取得最终期望的抗体之前花费时间和劳动等大量问题。作为克服免疫耐受的问题的技术,开发了不利用生物体内免疫的方法。该方法是在噬菌体粒子中埋入由各种抗体的可变区构成的单链抗体(singlechainvaluablefragment:scFv)基因,使单链抗体基因产物在噬菌体上展示,从利用该噬菌体所得到的单链抗体文库取得与目的抗原具有亲和性的克隆的方法,被称为噬菌体展示法。该技术在避免免疫耐受方面是优异的,但是由于从噬菌体文库得到的克隆是单链的,因此需要通过重组DNA技术等制备由双链构成的完全型的抗体。此外,在将单链抗体制成完全型抗体的过程中亲和性也会产生变化,需要调整可变区序列的情况也不少。因此,在时间和劳动方面,与利用生物体内免疫的方法相比,很难说特别地有进展。作为克服上述已知的单克隆抗体制造技术的课题的方法,报告了ADLib系统(专利文献1和非专利文献1)和对ADLib系统进一步进行改良的方法(专利文献2)。ADLib系统是,能够简便制备对所有类型的抗原具有期望的结合特性的多种抗体的技术。该方法是从利用自主进行抗体基因的多样化的来源于鸡B细胞的细胞株DT40构建的抗体文库选择性取得期望的抗体的技术。ADLib系统在体外系统抗体制造技术的优点即能够回避免疫耐受的方面,在迅速得到IgM型的完全抗体方面,与现有技术相比是优异的。通常,将抗体作为医药品对动物、人给药时,考虑到将体内的免疫原性最小化,要求与其动物种的类型匹配。因此,利用DNA重组技术制造将由小鼠等制造的单克隆抗体的恒定区取代为人型的嵌合抗体、将抗体的抗原识别部位(complementaritydeterminingregion:CDR)移植入人抗体的人化抗体。但是,在嵌合化·人化的过程中亲和性、功能也产生变化,能够制造保持功能的人化抗体的概率并非很高。此外,由于这些嵌合抗体或人化抗体来源于其他动物种的区域少,虽然与原单克隆抗体相比免疫原性低,但是由于抗原识别部位具有免疫原性,因此也存在出现抗抗体的问题。因此,尝试对小鼠基因导入人抗体基因来制造产生人抗体的动物。专利文献3中,将含有人抗体基因的染色体片段的微细胞移植入小鼠ES细胞,由该ES细胞制造嵌合小鼠,由此制造表达人抗体的嵌合小鼠。此外,非专利文献2中,通过将小鼠抗体基因座的基因组DNA取代为对应的人抗体基因座的基因组DNA,制造表达人抗体的嵌合小鼠。在任意的方法中,虽然均能够取得具有完全来源于人抗体基因的抗原识别部位的抗体,但是由于仍然利用了生物体内免疫,因此,在取得最终期望的抗体之前仍存在需要时间和劳动的问题。以上这类状况中,即使是使用ADLib系统的情形,为了制备抗体医药,需要由禽B细胞产生具有来源于人的氨基酸序列的抗体的方法。此外,为了进行各种疾病的治疗,需要简便产生具有多种抗原识别特性的抗体组的技术。但是,与将人抗体基因导入至小鼠进行“人化”的情形不同,将禽B细胞“人化”时,抗体的多样化系统的不同成为问题。因为在禽、兔子、牛、羊等动物的情形中,在抗体可变区的上游,存在与可变区类似的序列且其自身不作为抗体基因表达的被称为假基因的序列集中的区域,通过被称为基因转变的现象可变区序列的全部或一部分被假基因序列改写,从而产生多种序列。在人、小鼠等动物中,通过V(D)J重组引起抗体的多样化,而在禽、兔子、牛、羊等动物中,通过基因转变这一完全不同的机理引起抗体的多样化。此外,假基因区域有时成为含有非常大量的重复序列的结构,该区域的序列解析非常困难,即使在2015年3月的时间点也不能解码禽抗体重链假基因区域。由于这样的机理的不同、抗体基因座的结构上的问题,因此认为由禽B细胞构建产生期望的人抗体的系统是非常困难的。在这样的背景下,报告了制造将存在于DT40细胞的抗体基因座的可变区和假基因区域取代为来源于人的序列的细胞株(非专利文献3)。该文献中,将禽抗体可变区取代为含有属于荧光蛋白质的绿色荧光蛋白质(GFP)和青色荧光蛋白质(CFP)以及属于重组酶识别部位的attP序列的基因序列后,利用重组酶制造插入人抗体可变区和后述的假基因序列的DT40细胞,确认发生基因转变(非专利文献3、4)。但是,由于恒定区直接利用来源于禽的序列,因此表达的抗体分子是人-禽的嵌合抗体,抗体的免疫原性非常高。此外,由于轻链成为来源于人的κ链与来源于禽的λ链的融合蛋白,因此该抗体分子与天然的人抗体大大不同。因此,在制造人的治疗用抗体中需要进一步的嵌合化的操作,在嵌合化·人化的过程中仍然存在亲和性、功能产生变化的可能性。此外,此处使用的假基因区域的序列,关于轻链,利用了将插入的人抗体可变区的各个CDR序列每个CDR为1个氨基酸地取代为Tyr或Trp的人工序列的集合体即minimalistlibrary、对天然存在的人抗体可变区的变体在框架区实施变异的序列,关于重链,由将插入的人抗体可变区的各个CDR序列每个CDR为1个氨基酸地取代为Tyr或Trp的人工序列和其变异序列构成。这些序列是以利用少的假基因应对多种抗原作为目的而设计的序列,但是关于该细胞中的抗原特异性的抗体的取得没有报告。现有技术文献专利文献专利文献1:日本特许第4214234号专利文献2:WO2008/047480专利文献3:WO97/07671非专利文献非专利文献1:Seoetal,NatureBiotech,Volume23,pp731-735,2005非专利文献2:RecombinantAntibodiesforImmunotherapy,pp100-108,2009,CambridgePress非专利文献3:Schusseretal,PLOSONE,Volume8issue11e80108,2013非专利文献4:Leightonetal,FrontiersinImmunology,Volume6Article126,2015技术实现要素:鉴于上述事实,本发明的目的在于提供产生多种人抗体的禽B细胞。此外,本发明的目的在于提供从该禽B细胞产生抗体的方法。发明人等对产生多种人抗体的细胞的制造进行深入研究,最终在禽B细胞的抗体轻链和重链基因座中,插入或取代人抗体的轻链和重链的可变区和恒定区的基因后,成功取得表达人抗体的转化细胞。此外,发明人等进一步制造了在轻链·重链中插入多个来源于人的抗体可变区序列作为假基因的转化细胞,并将该细胞进行HDAC抑制剂处理后,确认与原来的禽B细胞同样地引起基因转变。此外,发明人等成功实现从该细胞产生抗原特异性的人抗体。即,发明人等确认通过制备上述细胞,从该细胞能够取得可变区进行各种改变的抗体,成功产生人抗体且多种抗体,从而完成本发明。即,本发明是以下的(1)~(21)。(1)一种禽B细胞,在抗体轻链基因座中,插入来源于人抗体轻链可变区和人抗体轻链恒定区的DNA序列的全部或一部分,或用来源于人抗体轻链可变区和人抗体轻链恒定区的DNA序列的全部或一部分取代,以及,在抗体重链基因座中,插入来源于人抗体重链可变区和人抗体重链恒定区的DNA序列的全部或一部分,或用来源于人抗体重链可变区和人抗体重链恒定区DNA序列的全部或一部分取代,以及,在抗体轻链假基因座中,插入2个以上来源于人抗体轻链可变区的DNA序列,或用2个以上来源于人抗体轻链可变区的DNA序列取代,和/或,在抗体重链假基因座中,插入2个以上来源于人抗体重链可变区的DNA序列,或用2个以上来源于人抗体重链可变区的DNA序列取代。(2)根据上述(1)记载的禽B细胞,其特征在于,在抗体轻链假基因座中插入的或者取代该抗体轻链假基因座的序列的、来源于人抗体轻链可变区的DNA序列的数量是5个以上。(3)根据上述(1)记载的禽B细胞,其特征在于,在抗体重链假基因座中插入的或取代该抗体重链假基因座的序列的、来源于人抗体重链可变区的DNA序列数量是5个以上。(4)根据上述(1)记载的禽B细胞,其特征在于,在抗体轻链假基因座中插入的或取代该抗体轻链假基因座的序列的、来源于人抗体轻链可变区的DNA序列的数量是15个以上。(5)根据上述(1)记载的禽B细胞,其特征在于,在抗体重链假基因座中插入的或取代该抗体重链假基因座的序列的、来源于人抗体重链可变区的DNA序列的数量是15个以上。(6)根据上述(1)~(5)中任一项记载的禽B细胞,其特征在于,在抗体轻链基因座中插入的来源于人抗体轻链可变区的DNA序列的位置位于来源于人抗体轻链恒定区的DNA序列所存在的位置的上游,以及在抗体重链基因座中插入的来源于人抗体重链可变区的DNA序列的位置位于来源于人抗体重链恒定区的DNA序列所存在的位置的上游。(7)根据上述(6)记载的禽B细胞,其特征在于,在抗体轻链基因座中插入的来源于人抗体轻链可变区的DNA序列和来源于恒定区的DNA序列的位置与禽抗体轻链可变区和恒定区的位置相比位于上游,以及在抗体重链基因座中插入的人抗体重链可变区和恒定区的位置与禽抗体重链可变区和恒定区的位置相比位于上游。(8)根据上述(1)~(7)中任一项记载的禽B细胞,其特征在于,上述人抗体重链是γ链。(9)根据上述(1)~(8)中任一项记载的禽B细胞,其特征在于,上述人抗体轻链是λ链或κ链。(10)根据上述(1)~(9)中任一项记载的禽B细胞,其特征在于,上述禽B细胞具有在细胞表面表达人抗体且还在培养液中分泌人抗体的能力。(11)根据上述(1)~(10)中任一项记载的禽B细胞,其特征在于,禽B细胞是DT40细胞。(12)根据上述(1)~(11)中任一项记载的禽B细胞,进行使染色质松弛的处理。(13)根据上述(12)记载的禽B细胞,其中,上述使染色质松弛的处理是使该禽B细胞中的组蛋白去乙酰化酶的功能降低或丧失。(14)根据上述(13)记载的禽B细胞,其中,上述使组蛋白去乙酰化酶的功能降低或丧失的方法是使该禽B细胞中的组蛋白去乙酰化酶基因的表达降低或丧失。(15)根据上述(14)记载的禽B细胞,其特征在于,上述组蛋白去乙酰化酶是HDAC2。(16)根据上述(13)记载的禽B细胞,其特征在于,上述使组蛋白去乙酰化酶的功能降低或丧失的方法是利用HDAC抑制剂处理。(17)由上述(12)~(16)中任一项记载的禽B细胞构成的产生抗体的细胞文库。(18)由上述(1)~(16)中任一项记载的禽B细胞制造抗体的方法。(19)通过上述(18)记载的方法取得的抗体。(20)用于制造上述(1)~(16)中任一项记载的禽B细胞的试剂盒。(21)用于通过上述(18)记载的方法产生抗体的试剂盒。由本发明的细胞,能够简便且迅速地取得识别多种抗原的人抗体。附图说明图1是示意性地表示用于将DT40细胞的抗体轻链恒定区和抗体轻链可变区用来源于人的抗体(IgG)轻链恒定区和抗体轻链可变区进行取代的靶向载体的结构的图。cpV1~cpV3:禽假基因图2是示意性地表示用于在DT40细胞的抗体轻链假基因区域下游,插入基因转变确认用序列(GC确认序列;由来源于人抗体可变区的序列构成的人假基因。以下相同)的靶向载体的结构的图。cpV1~cpV3:禽假基因图3是示意性地表示用于DT40细胞的抗体轻链假基因区域的缺失、和将DT40细胞的抗体轻链假基因区域用GC确认序列进行取代的靶向载体的结构的图。cpV25、cpV1:禽假基因图4是示意性地表示用于将DT40细胞的抗体重链恒定区和抗体可变区用来源于人的抗体(IgG)重链恒定区和抗体重链可变区进行取代,进一步插入GC确认序列的靶向载体的结构的图。图5是示意性地表示将DT40细胞的抗体轻链基因区域用来源于人的抗体轻链可变区和抗体轻链恒定区取代的顺序,和得到的DT40细胞的抗体轻链基因区域的图。cpV1~cpV3:禽假基因图6是示意性地表示用于将DT40细胞的抗体轻链基因区域用来源于人的抗体轻链可变区和抗体轻链恒定区基因取代,进一步在DT40细胞的抗体轻链假基因区域的下游插入GC确认序列的顺序,和得到的DT40细胞的抗体轻链基因区域的图。cpV1~cpV3:禽假基因、hpV1~hpV2:人假基因图7是示意性地表示用于将DT40细胞的抗体轻链基因区域用来源于人的抗体轻链可变区和抗体轻链恒定区基因取代,进一步使DT40细胞的抗体轻链假基因区域缺失的顺序,和得到的DT40细胞的抗体轻链基因区域的图。cpV25、cpV1:禽假基因图8是示意性地表示用于将DT40细胞的抗体轻链基因区域用来源于人的抗体轻链可变区和抗体轻链恒定区基因取代,进一步将DT40细胞的假基因区域取代为GC确认序列的顺序,和得到的DT40细胞抗体轻链基因区域的图。cpV25、cpV1:禽假基因、hpV1、hpV2:人假基因图9表示对在DT40细胞的抗体轻链基因区域插入来源于人的抗体轻链基因区域的细胞株、或将DT40细胞的抗体轻链基因区域用人抗体轻链基因取代的细胞株产生的抗体,利用流式细胞仪进行解析的结果。图10是示意性地表示将DT40细胞的抗体重链基因区域变换为来源于人的抗体重链可变区和抗体重链恒定区的顺序,和得到的DT40细胞的抗体重链基因区域的图。图11是示意性地表示用于将DT40细胞的抗体重链基因区域变换为来源于人的抗体重链可变区和抗体重链恒定区基因,进一步在DT40细胞的重链假基因区域的下游插入GC确认序列的顺序,和得到的DT40细胞的抗体轻链基因区域、以及使用的靶向载体的结构的图。hpV1、hpV2:人假基因图12表示针对将DT40细胞的抗体重链基因区域变换为来源于人的抗体重链可变区和抗体重链恒定区基因、进一步在DT40细胞的重链假基因区域的下游插入GC确认序列的细胞株所产生的抗体,利用流式细胞仪进行解析的结果。图13是示意性地表示将DT40细胞的抗体轻链的假基因区域用由来源于人Igλ的可变区的序列构成的假基因(人轻链假基因)进行取代的顺序,和得到的DT40细胞的抗体轻链区域,以及使用的靶向载体的结构的图。图14是示意性地表示用于将DT40细胞的抗体重链恒定区取代为人IgG1的重链基因区域的靶向载体的结构的图。图15是示意性地表示用于在DT40细胞的抗体重链可变区导入来源于人的抗体重链可变区和5个由来源于人Igγ的可变区的序列构成的假基因(人重链假基因)的靶向载体的结构的图。图16是示意性地表示将DT40细胞的抗体轻链基因区域用来源于人的抗体轻链可变区和抗体轻链恒定区取代并插入人轻链假基因序列(5个)的顺序,和得到的DT40细胞抗体轻链基因区域的图。cpV25、cpV1:禽假基因图17是示意性地表示在DT40细胞的抗体重链基因区域导入来源于人的抗体重链可变区和抗体重链恒定区并插入人重链假基因序列(5个)的顺序,和得到的DT40细胞抗体重链基因区域的图。图18表示针对将DT40细胞的抗体轻链基因区域和抗体重链基因区域变换为来源于人的抗体基因并插入人轻链假基因序列(5个)的细胞株所产生的抗体,利用流式细胞仪进行解析的结果。图19是示意性地表示将DT40细胞的抗体轻链基因区域用来源于人的抗体轻链可变区和抗体轻链恒定区取代并插入人重链假基因序列(15个)的顺序,和使用的靶向载体的结构的图。pV25、pV1:禽假基因图20是示意性地表示在DT40细胞的抗体重链基因区域导入来源于人的抗体重链可变区和抗体重链恒定区并插入人重链假基因序列(15个)的顺序,和使用的靶向载体的结构的图。图21是针对将DT40细胞的抗体轻链基因区域和抗体重链基因区域变换为来源于人的抗体基因区域并插入人轻链假基因序列(15个)的L15/H15细胞株所产生的抗体,利用流式细胞仪进行解析的结果。图22表示对于图21表示的结果,将存在错误的数据基于优先权日前的数据进行修正的结果。图23是示意性地表示将DT40细胞的抗体重链可变区用来源于人的抗体重链可变区取代的同时在DT40细胞的抗体重链可变区中插入RMCE用盒序列的顺序,和使用的靶向载体的结构的图。图24是示意性地表示在DT40细胞的抗体重链基因区域中插入30个人重链假基因序列的顺序,和使用的靶向载体的结构的图。图25表示针对将DT40细胞的抗体轻链基因区域和抗体重链基因区域变换为来源于人的抗体基因区域并在轻链插入15个人轻链假基因序列、在重链插入30个人重链假基因序列的L15/H30细胞株所产生的抗体,利用流式细胞仪进行解析的结果。图26是示意性地表示在DT40细胞的抗体轻链基因区域中插入30个人轻链假基因序列的顺序,和使用的靶向载体的结构的图。图27表示针对将DT40细胞的抗体轻链基因区域和抗体重链基因区域变换为来源于人的抗体基因区域并在轻链插入30个人轻链假基因序列、在重链插入30个人重链假基因序列的L30/H30细胞株所产生的抗体,利用流式细胞仪进行解析的结果。图28表示针对将DT40细胞的抗体轻链基因区域和抗体重链基因区域变换为来源于人的抗体基因区域并在轻链插入30个人轻链假基因序列、在重链插入15个人重链假基因序列的L30/H15细胞株所产生的抗体,利用流式细胞仪进行解析的结果。图29是示意性地表示利用RMCE法在L30/H15细胞株的抗体重链基因区域中在正向或反向追加插入15个人重链假基因序列而合计为30个的顺序,和使用的靶向载体的结构的图。图中,for和rev分别是指正向、反向。以下,其他图中相同。图30表示针对利用RMCE法在L30/H15细胞株的抗体重链基因区域中在正向或反向追加插入15个人重链假基因序列的细胞株(正向为L30/H15f15f细胞株,反向为L30/H15r15f细胞株)所产生的抗体,利用流式细胞仪进行解析的结果。图31是示意性地表示在L30/H15f15f细胞株和L30/H15r15f细胞株的抗体重链基因区域中分别在正向或反向追加插入15个人重链假基因序列而合计为45个的顺序,和使用的靶向载体的结构的图。图32表示针对在L30/H15f15f细胞株的抗体重链基因区域中在正向追加插入15个人重链假基因序列的细胞株(L30/H15f15f15f细胞株)所产生的抗体,利用流式细胞仪进行解析的结果。图33表示针对在L30/H15r15f细胞株的抗体重链基因区域中在反向追加插入15个人重链假基因序列的细胞株(L30/H15r15r15f细胞株)所产生的抗体,利用流式细胞仪进行解析的结果。图34是示意性地表示利用RMCE法在L30/H15f15f15f细胞株和L30/H15r15r15f细胞株的抗体重链基因区域中分别在正向或反向追加插入15个人重链假基因序列而合计为60个的顺序,和使用的靶向载体的结构的图。图35表示针对在DT40细胞的抗体轻链基因区域中插入30个人轻链假基因序列、在抗体重链基因区域中插入60个人重链假基因的细胞株所产生的抗体,利用流式细胞仪进行解析的结果。图36表示由TSA处理的L15/H15细胞株,使用ADLib系统分离与PlexinA4蛋白质结合的细胞团,利用流式细胞仪进行解析的结果。图37表示对图36的P5区的细胞团进行单细胞分选(singlecellsorting)后,在96孔板上播种培养,对各孔的细胞上清进行ELISA解析的结果。横轴表示作为测定样品的细胞上清所来源的96孔板的各孔划分的编号。Trypsininhibitor:胰蛋白酶抑制剂、SA:链霉亲和素、OA:卵清蛋白图38表示利用蛋白质印迹法(WesternBlot),对阳性克隆的#62和#20分泌的抗体进行分析。在还原和非还原的条件下处理样品,使用山羊抗人IgGγ抗体进行蛋白质印迹(WesternBlotting)的结果。图39表示利用蛋白质印迹法,对由阳性克隆#62和#20分泌的抗体进行分析。在还原和非还原的条件下处理样品,使用山羊抗人IgGλ抗体进行蛋白质印迹的结果。图40表示利用蛋白质印迹法,对由阳性克隆#62和#20分泌的抗体进行分析。在还原和非还原的条件下处理样品,使用山羊抗禽IgM抗体进行蛋白质印迹的结果。图41表示由TSA处理的L15/H15细胞株,使用ADLib系统对与His-AP-hSema3A结合的细胞团进行1次筛选的结果。图42表示将与His-AP-hSema3A结合的细胞团单克隆化之后进行的2次筛选的结果。图43表示利用蛋白质印迹法,对由2次筛选后的阳性克隆分泌的His-AP-hSema3A抗体进行分析。在还原和非还原的条件下处理样品,使用山羊抗人IgGγ抗体或山羊抗人IgGλ抗体进行蛋白质印迹的结果。图44表示由TSA处理的L30/H45细胞株,使用ADLib系统对与IL-8结合的细胞团进行1次筛选的结果。图45表示在将与IL-8结合的细胞团单克隆化之后进行的2次筛选的结果。图46表示利用蛋白质印迹法,对由2次筛选后的阳性克隆分泌的IL-8抗体进行分析。在还原和非还原的条件下处理样品,使用山羊抗人IgGγ抗体或山羊抗人IgGλ抗体进行蛋白质印迹的结果。具体实施方式本发明的实施方式之一是禽B细胞,在抗体轻链基因座中,插入来源于人抗体轻链可变区和人抗体轻链恒定区的DNA序列的全部或一部分,或用来源于人抗体轻链可变区和人抗体轻链恒定区的DNA序列的全部或一部分取代,以及,在抗体重链基因座中,插入来源于人抗体重链可变区和人抗体重链恒定区的DNA序列的全部或一部分,或用来源于的人抗体重链可变区和人抗体重链恒定区的DNA序列的全部或一部分取代,以及,在抗体轻链假基因座中,插入2个以上来源于人抗体轻链可变区的DNA序列,或用2个以上来源于人抗体轻链可变区的DNA序列取代,和/或,在抗体重链假基因座中,插入2个以上来源于人抗体重链可变区的DNA序列,或用2个以上来源于人抗体重链可变区的DNA序列取代。本发明的B细胞是来源于禽的产生抗体的B细胞,例如,能够举出来源于鸡的DT40细胞。除此以外,也能够使用具有通过基因转变使抗体序列多样化的系统的牛、羊、兔子等的B细胞。此外,本发明的禽B细胞,在其抗体轻链基因座中,插入来源于人抗体轻链可变区和人抗体轻链恒定区的DNA序列的全部或一部分,或用来源于人抗体轻链可变区和人抗体轻链恒定区的DNA序列的全部或一部分取代,以及,在其抗体重链基因座中,插入来源于人抗体重链可变区和人抗体重链恒定区的DNA序列的全部或一部分,或用来源于人抗体重链可变区和人抗体重链恒定区的DNA序列的全部或一部分取代,以及,在抗体轻链假基因座中,插入2个以上来源于人抗体轻链可变区的DNA序列,或用2个以上来源于人抗体轻链可变区的DNA序列取代,和/或,在抗体重链假基因座中,插入2个以上来源于人抗体重链可变区的DNA序列,或用2个以上来源于人抗体重链可变区的DNA序列取代,除此之外,还可以对其他基因区域等实施变异、修饰。此处,抗体轻链基因座是存在编码抗体的轻链可变区和轻链恒定区的基因的基因座,抗体重链基因座是存在编码抗体的重链可变区和重链恒定区的基因的基因座。此外,假基因是虽然具有与发挥功能的基因类似的序列但不作为表达的基因发挥功能的DNA序列。禽的抗体轻链假基因座和重链假基因座分别与存在功能基因的抗体轻链基因座和重链基因座相比位于上游,通过基因转变产生多样性。在人的基因组中的抗体基因座不存在假基因,但在本说明书中,将插入的人抗体可变区和出于产生基因转变的目的而导入禽抗体基因座中的、具有与人抗体可变区类似的序列的DNA序列统称为“人假基因”。在禽抗体轻链基因座中插入的、或与禽抗体轻链基因座的序列替换的来源于人抗体轻链可变区和人抗体轻链恒定区的DNA序列分别可以是人抗体轻链可变区和人抗体轻链恒定区的全部序列,或者,也可以是其部分序列。此外,在禽抗体轻链基因座中插入的来源于人抗体轻链可变区的DNA序列的位置,优选为来源于人抗体轻链恒定区的DNA序列的插入位置的上游。同样,在禽抗体重链基因座中插入的、或与禽抗体重链基因座的序列替换的来源于人抗体重链可变区和人抗体恒定区的DNA序列分别可以是人重链可变区和重链恒定区的全部序列,或者,也可以是其部分序列。此外,在禽抗体重链基因座中插入的来源于人抗体重链可变区的DNA序列的位置,优选为来源于人抗体重链恒定区的DNA序列的插入位置的上游。此外,优选在禽抗体轻链基因座中插入的来源于人抗体轻链可变区和人抗体轻链恒定区的DNA序列的位置,与禽抗体轻链可变区和禽抗体轻链恒定区的位置相比位于上游。同样,优选在禽抗体重链基因座中插入的来源于人抗体重链可变区和重链恒定区的DNA序列的位置,与禽抗体重链可变区和重链恒定区的位置相比位于上游。本实施方式中,在禽抗体轻链假基因座中插入的、或与禽抗体轻链假基因座的DNA序列替换的来源于人抗体轻链可变区的DNA序列(本说明书和附图中,也记载为“人轻链假基因序列”),可以使用公知的人抗体轻链可变区的序列。这样的序列信息,例如能够从VBase(http://www2.mrc-lmb.cam.ac.uk/vbase/list2.php)这样的网站获得。该人轻链假基因序列可以使用含有人抗体轻链可变区的CDR1、CDR2、CDR3的全部或者其一部分的DNA序列,但是优选各CDR不同于在禽抗体轻链基因座中插入的或与禽抗体轻链基因座的序列替换的人抗体轻链可变区的DNA序列、其他的人轻链假基因序列的CDR。此外,关于人轻链假基因序列的框架(FW)区,优选跟在禽抗体轻链基因座中插入的或与禽抗体轻链基因的序列替换的人抗体轻链可变区的框架区一致。例如,作为抗体轻链可变区选择序列号5的DNA序列时,轻链假基因可以将序列号69~序列号88和序列号127~序列号141所示的序列等组合使用。同样,在禽抗体重链假基因座中插入的、或者与禽抗体重链假基因座的序列替换的来源于人抗体重链可变区的DNA序列(本说明书和附图中,也记载为“人重链假基因序列”),可以使用公知的人抗体重链可变区的序列。这样的序列信息,例如可以从VBase(http://www2.mrc-lmb.cam.ac.uk/vbase/list2.php)这样的网站获得。该人重链假基因序列可以使用包括人抗体重链可变区的CDR1、CDR2、CDR3的全部或其一部分的DNA序列,但优选各CDR不同于在禽抗体重链基因座中插入的或者与禽抗体重链基因座的序列替换的人抗体重链可变区的DNA序列、其他人重链假基因序列的CDR。此外,关于人重链假基因序列的框架(FW)区,优选跟在禽重链抗体基因座中插入的、或与禽重链抗体基因座的序列替换的人抗体重链可变区的框架区一致。例如,作为抗体重链可变区选择序列号30的DNA序列时,重链假基因可以将序列号89~序列号108、序列号127~序列号141、序列号147~序列号161和序列号165~序列号179所示的序列等组合使用。本实施方式中,在禽抗体轻链假基因座中插入的来源于人抗体轻链可变区的DNA序列的位置,优选与在禽抗体轻链基因座中插入的人抗体轻链可变区相比位于上游,此外,在残留禽抗体轻链假基因序列时,优选与禽抗体轻链假基因区域相比位于下游。同样,在禽抗体重链假基因座中插入的来源于人抗体重链可变区的DNA序列的位置,优选与在禽抗体重链基因座中插入的人抗体重链可变区相比位于上游,此外,在残留禽抗体重链假基因序列时,优选与禽抗体重链假基因区域相比位于下游。此外,本实施方式中,在禽抗体可变区的上游作为假基因插入的或取代的来源于人抗体可变区的DNA序列,与在禽抗体可变区插入或取代的人抗体可变区序列可以是同向也可以是反向,均能够利用。在禽抗体轻链假基因座中插入的、或取代(替换)禽抗体轻链假基因座的假基因的来源于人抗体轻链可变区的DNA序列的数量,优选为2个以上,更优选为5个以上、7个以上、9个以上、11个以上、13个以上,进一步优选为15个以上、20个以上、25个以上。同样,在禽抗体重链假基因座中插入的、或取代(替换)禽抗体重链假基因座的假基因的来源于人抗体重链可变区的DNA序列的数量,优选为2个以上,更优选为5个以上、7个以上、9个以上、11个以上、13个以上,进一步优选为15个以上、20个以上、25个以上,最优选为30个以上。应予说明,此处所述的来源于人抗体轻链可变区的DNA序列或来源于人抗体重链可变区的DNA序列,具有FW1、CDR1、FW2、CDR2、FW3、CDR3、FW4中的任意1个,以遍及可变区的区域所含有的连续的一系列的可变区相同序列作为1个进行处理。本实施方式中,在禽抗体重链基因座中插入的人抗体重链的种类,可以是γ链、s链、α链、δ链、ε链中的任一个,优选为γ链。此外,在禽抗体轻链基因座和禽抗体轻链假基因座中插入的人抗体轻链的种类,可以是λ链和κ链的任一个。本实施方式中,为了将进行了基因导入的禽B细胞作为抗体文库利用,优选该禽B细胞具有不仅在细胞膜表面表达人抗体,而且在培养液中分泌人抗体的能力。因此,转录导入的人抗体基因时,需要适当引起与生物体中的表达控制的机理类似的选择性剪接(alternativesplicing)。因此,在恒定区的基因导入中,理想的是利用与禽抗体重链恒定区完全相同的基因组结构,但是由于禽的该基因组区域的碱基序列在2015年3月这一时间点没有被解读,因此不能使用该序列。本实施方式中利用小鼠抗体重链基因组序列中的内含子序列,最终,成功实现同时表达细胞膜表面和分泌型两种的人抗体,因此,也能用来源于小鼠、人等公知哺乳动物的内含子序列代替使用。本实施方式中,作为在禽抗体基因座中插入人抗体基因和/或用人抗体基因取代的方法,使用利用了同源重组的敲入法。此外,在禽抗体链基因座的假基因区域中插入来源于人抗体可变区的序列和/或用人抗体基因取代的方法,除了利用同源重组的敲入法之外,还优选使用利用了部位特异性重组的重组酶介导的盒式交换(Recombinasemediatedcassetteexchange,RMCE)法。本实施方式制造的禽B细胞中,除了如上述所示地插入来源于人的抗体基因座的DNA序列或用来源于人的抗体基因座的DNA序列取代的细胞之外,也包括在该细胞的来源于人的抗体轻链或重链可变区进一步导入多种多样的变异而成的禽B细胞。抗体的多样性可以通过将可变区重新编辑,产生多种可变区的序列从而实现。因此,本实施方式制造的禽B细胞中,也包括实施了导入对可变区的重新编辑必要的多种变异的处理而成的禽B细胞。此处,导入对可变区的重新编辑必要的变异的方法,可利用例如使用使XRCC2和XRCC3缺失的B细胞的方法(例如,Cumberetal.,NatureBiotech.204:1129-11342002或日本特开2003-503750等)、控制AID基因的表达的方法(例如,Kanayamaetal.,NucleicAcidsRes.34:e10.,2006或日本特开2004-298072等),或者,使染色质松弛促进基因转变的方法(例如,专利文献1、专利文献2或非专利文献1等)等该
技术领域:
公知的方法。特别优选的方法是使染色质松弛促进基因转变的方法。作为使禽B细胞中的染色质松弛的方法,例如,可举出利用某种方法抑制禽B细胞中内在的组蛋白去乙酰化酶(HDAC)活性而显著促进该细胞内的基因转变的方法(详细内容参见上述专利文献1、专利文献2或非专利文献1等)。作为抑制细胞内在的HDAC的活性的方法,可使用利用HDAC抑制剂处理本发明的B细胞的方法(参见专利文献1或非专利文献1)、使禽B细胞中的HDAC基因的功能降低或丧失的方法(参见专利文献2)等。HDAC抑制剂是抑制HDAC的活性的抑制剂即可没有特别限制,例如,能够使用曲古柳菌素A(TSA)、丁酸、丙戊酸、辛二酰苯胺异羟肟酸(suberoylanilidehydroxamicacid,SAHA)、CBHA(间羧基肉桂酸双羟酰胺,m-carboxycinnamicacidbis-hydroxamide)、PTACH、Apicidin、Scriptaid、M344、APHAcompound8、BML-210、Oxamflatin、MS-275、等。此外,可能的情况下,也可以将活性抑制对象HDAC的失活型蛋白质(显性负效应)等作为抑制物质使用。使HDAC基因的功能降低或丧失时,成为使基因功能降低或丧失的对象的HDAC的同工酶,因使用的抗体产生B细胞不同而不同,优选为HDAC2基因(详细地,参见专利文献2)。与上述实施方式相关的本发明的实施方式中,包括由本发明的禽B细胞构成的产生抗体的细胞文库。该细胞文库是本发明的禽B细胞,是产生针对各种抗原的抗体的该禽B细胞的集团。本发明的上述实施方式记载的禽B细胞是通过在禽抗体假基因座插入来源于人的抗体可变区的DNA序列等而在产生的抗体可变区序列中产生多样性,而且通过染色质的松弛等处理,进一步使抗体可变区的序列多样化,具有能够针对各种抗原产生抗体的能力的细胞。因此,包含多种本发明的禽B细胞的集团能够称为针对多种抗原产生抗体的细胞团。本发明的其他实施方式是由本发明的禽B细胞产生抗体的方法,以及制备的抗体。由本发明的禽B细胞产生的抗体是人型抗体,更优选为人抗体。人型抗体是在产生的抗体的重链或轻链的氨基酸序列中含有一部分来源于禽基因的序列的抗体,人抗体是产生的抗体的重链或轻链的氨基酸序列全部为来源于人基因的序列的抗体。本发明的禽B细胞在适于该细胞的培养条件下进行培养时,产生人型抗体或人抗体。已知在DT40细胞这类来源于禽B细胞的细胞株中,虽然不具有那么高的效率,但是在抗体基因座中产生基因转变。因此,本发明的禽B细胞中,也产生基因转变,通过被插入来源于人的抗体基因的抗体基因座的基因转变,获得产生的抗体的第一次的多样性。进而,在此基础上,通过使本发明的禽B细胞的染色质松弛,抗体基因座中的基因转变效率进一步上升,从可制备能够产生更多种抗体的禽B细胞的集团。为了利用这样的染色质的松弛而选择产生显示期望的抗原特异性的抗体的禽B细胞,可利用ADLib系统。前述的产生抗体的细胞文库也能通过利用ADLib系统而制备(参见专利文献1或非专利文献1)。此外,特别是,作为取得针对膜结合蛋白质的期望的抗体的方法,可利用ADLibaxCELL(antigenexpressingcell)法(WO2010/064454)。本发明的另一实施方式包括用于制造本发明的禽B细胞的试剂盒以及用于产生抗体的试剂盒。作为用于制造禽B细胞的试剂盒,除了用于培养禽B细胞而必要的培养基、必要的添加剂等之外,还包括用于在禽B细胞的抗体基因座等中插入来源于人抗体基因的DNA序列或者用来源于人抗体基因的DNA序列进行取代的载体、试剂类。此外,还可以包括用于制备产生抗体的细胞文库所需要的要素,例如HDAC抑制剂、用于进行使HDAC基因的功能降低或功能丧失所需要的试剂。此外,用于产生抗体的试剂盒中,作为用于选择具有期望的抗原特异性的抗体的试剂类,例如,除了各种抗原、磁珠等之外,还可以含有磁珠制备用试剂、抗体选择所使用的标记抗体、ELISA等检查所需要的板等,如果认为抗体制造中需要的物质则可以含有任意的物质。以下示出实施例做进一步详细说明,本发明不受到实施例的任何限制。实施例基因转变的确认1.载体的制备1-1.禽抗体轻链(Igλ)的恒定区·可变区的取代(humanLCKIvector)为了取代禽Igλ的恒定区和可变区,制造如图1所示的靶向载体。以下示出各个部分的制造方法。(1)人Igλ的可变区和恒定区将由人伯基特淋巴瘤细胞株Ramos制备的cDNA作为模板,通过使用正义引物(TCCACCATGGCCTGGGCTCTG)(序列号1)和反义引物(GTTGAGAACCTATGAACATTCTGTAGGGGCCAC)(序列号2)的PCR,扩增人抗体轻链(Igλ)的可变区和恒定区,克隆至pGEM-T-Easy(PromegaCorporation),得到pGEM-hIgG1-LC。将该质粒pGEM-hIgG1-LC作为模板,通过使用正义引物(ATTGGCGCGCCTCTCCAGGTTCCCTGGTGCAGGCACAGTCTGCCCTGACTCAGC)(序列号3)和反义引物(TTCCATATGAGCGACTCACCTAGGACGGTCAGCTTGGTCC)(序列号4)的PCR,得到人Igλ的可变区(序列号5)。此外,将pGEM-hIgG1-LC作为模板,通过使用正义引物(ATTGGCGCGCCTCTGCCTCTCTCTTGCAGGTCAGCCCAAGGCTGCCCCCTC)(序列号6)和反义引物(GGAATTCCATATGGAGTGGGACTACTATGAACATTCTGTAGGGG)(序列号7)的PCR,得到人Igλ的恒定区(序列号8)。(2)左臂(LeftArm)将鸡B细胞株DT40的基因组DNA作为模板,通过使用正义引物(GAGATCTCCTCCTCCCATCC)(序列号9)和反义引物(CAAAGGACACGACAGAGCAA)(序列号10)的PCR,扩增从禽Igλ的可变区上游至恒定区下游的区域。通过电泳分离为Functionalallele(功能性等位基因)和Non-functionalallele(非功能性等位基因),切出约8.0kbp的功能性等位基因,克隆至pGEM-T-Easy,得到pGEM-cIgM-LCgenome。将pGEM-cIgM-LCgenome作为模板,通过使用正义引物(TGTCTCGAGTGAAGGTCACCAAGGATGG))(序列号11)和反义引物(TTAAGCTTGGAGAGGAGAGAGGGGAGAA)(序列号12)的PCR,得到序列号13表示的左臂。(3)中臂(CenterArm)通过pGEM-cIgM-LCgenome的序列解析确定位于禽Igλ的可变区和恒定区之间的内含子区域的序列,合成序列号14表示的中臂(在中途插入两端具有loxP序列的杀稻瘟菌素耐药性基因)。(4)右臂(RightArm)使用pGEM-cIgM-LCgenome确定禽Igλ的恒定区的下游区域的序列,合成序列号15表示的右臂(在中途插入两端具有lox2272序列的新霉素耐药性基因)。(5)靶向载体的构建在pBluescript中以成为图1表示的结构的方式组装上述左臂、人Igλ可变区、中臂、人Igλ恒定区、右臂,得到humanLCKIvector。该载体的基因序列示于序列号16。1-2.对禽抗体轻链(Igλ)的假基因区域下游的敲入(KI-C-INvector)为了在禽Igλ的假基因区域下游插入用于确认基因转变的序列(GC确认序列;由来源于人的Igλ的可变区的序列构成的人假基因)(序列号17),首先制造如图2所示的靶向载体。各个部分的制造方法如以下所示。(1)左臂将DT40的基因组DNA作为模板,使用正义引物(TTCTGTGAGCTGAGAAAAGGAGTGTA)(序列号18)和反义引物(CCTGCATTGTGGCACAGCGGGGTT)(序列号19),利用PCR将可变区的启动子的上游的4.3kb(假基因(pV)1~3)扩增后,克隆至pCR4blunt-TOPO(LifeTechnologiesCorporation),得到pCR4-cIgM-LCpV1genome。基于该序列,克隆包含pV1~3的区域(序列号20),作为左臂。(2)右臂基于上一项得到的pCR4-cIgM-LCpV1genome的序列,克隆相当于pV1与启动子之间的区域(序列号21)的部分,作为右臂。(3)靶向载体的构建在pCR4blunt-TOPO中以成为图2表示的结构的方式组装上述左臂、新霉素耐药性基因、右臂,得到KI-C-INvector。该载体的基因序列示于序列号22。1-3.在禽抗体轻链(Igλ)的假基因区域下游插入GC确认序列(KI-C-IN-Cvector)为了在禽Igλ的假基因区域下游插入GC确认序列,制造如图2所示的靶向载体。各个部分的制造方法如以下所示。(1)GC确认用序列基于上述1-1中克隆的人Igλ的可变区,设计在各个CDR中分别插入3个碱基的变异序列和分别缺失3个碱基的变异序列,合成将其用禽Igλ假基因区域的非翻译区域序列连接的GC确认用序列(序列号17)。(2)靶向载体的构建在上述1-2中制造的KI-C-INvector中,以成为图2表示的结构的方式组装GC确认用序列,得到KI-C-IN-Cvector。该载体的基因序列示于序列号23。1-4.禽抗体轻链(Igλ)的假基因区域的缺失(KI-C-DEvector)为了使禽Igλ的假基因区域缺失,制造图3所示的靶向载体。各个部分的制造方法如以下所示。(1)左臂将DT40的基因组DNA作为模板,使用正义引物(CGCTTTGTACGAACGTTGTCACGT)(序列号24)和反义引物(TACCTGAAGGTCTCTTTGTGTTTTG)(序列号25),利用PCR扩增pV25的上游的约3.0kb后,克隆至pCR4blunt-TOPO,得到序列号26表示的左臂。(2)右臂使用与上述1-2中制备的右臂相同的右臂。(3)靶向载体的构建在pCR4blunt-TOPO中以成为图3表示的结构的方式组装上述的左臂、新霉素耐药性基因、右臂,得到KI-C-DEvector。该载体的基因序列示于序列号27。1-5.将禽抗体轻链(Igλ)假基因区域取代为GC确认用序列(KI-C-DE-Cvector)为了将禽Igλ的假基因区域取代为GC确认用序列,制造图3所示的靶向载体。各个部分的制造方法如以下所示。(1)GC确认用序列使用上述1-2中制备的GC确认用序列。(2)靶向载体的构建在上述1-3中制造的KI-C-DEvector中,以成为图3表示的结构的方式组装上述的GC确认用序列,得到KI-C-DE-Cvector。该载体的基因序列示于序列号28。1-6.对禽抗体重链(IgM)的恒定区·可变区的敲入为了将从禽IgM(IgM)的可变区至恒定区μ1的区域取代为人抗体重链(IgG1),制造图4所示的靶向载体。各个部分的制造方法如以下所示。(1)人IgG1恒定区从含有人IgG1的人基因组染色体1432.33的利用鸟枪法测序法得到的序列中,取得相当于IgG1的序列(登录号:NW_001838121.1)。从第41520位碱基与第43117位碱基之间的序列提取相当于“IGH1”的”C_region”的CH1、hinge、CH2、CH3、transmembrane结构域序列,设计将内含子取代为来源于小鼠IgG2a的序列而得的序列,通过合成得到人IgG1恒定区序列(序列号29)。(2)人IgG1可变区基于来源于人的生殖细胞的IgG1可变区基因的序列,设计连接VH3-23、D5-12、JH1序列的序列,附加来源于禽的分泌信号序列、剪接信号序列,作为人IgG1可变区(序列号30)。(3)左臂将DT40的基因组DNA作为模板,使用正义引物(TTCCCGAAGCGAAAGCCGCGT)(序列号31)和反义引物(ACTCACCGGAGGAGACGATGA)(序列号32),利用PCR扩增从禽IgM可变区上游至可变区的区域,克隆约4.1kbp的片段,得到pCR4Blunt-TOPO-cIgM-HCpV1genome。基于该序列,设计·合成在3’端附加Vlox序列的左臂(序列号33)。(4)中臂1基于在上述的左臂克隆时克隆的从禽IgM可变区上游至可变区的约4.1kbp的序列,合成相当于禽IgM可变区上游的约1.4kbp的中臂1(序列号34)。(5)中臂2将DT40的基因组DNA作为模板,使用正义引物(GGGGATCCTGGGTCAGTCGAAGGGGGCG)(序列号35)和反义引物(GTGCGGCCGCCAAAAGCGGTAAAATCCACCC)(序列号36),利用PCR扩增禽IgM可变区下游的约4.3kbp之后进行克隆,得到序列号37表示的中臂2。(6)右臂将DT40的基因组DNA作为模板,使用正义引物(CCAAACCACCTCCTGGTGTCC)(序列号38)和反义引物(CAAACCAAAACTACGGATTCTCTGACC)(序列号39),利用PCR扩增含有禽IgM恒定区μ2的约0.9kbp之后进行克隆,得到序列号40表示的右臂。(7)靶向载体的构建在pBluescript中以成为图4表示的结构的方式组装上述的左臂、多克隆位点、中臂1、人IgG1可变区、杀稻瘟菌素耐药性基因、中臂2、人IgG1恒定区、新霉素耐药性基因、右臂,得到humanHCV-Cvector。该载体的基因序列示于序列号41。1-7.对禽抗体重链(IgM)的恒定区·可变区·假基因区域的敲入(humanHCKIvector)为了在禽IgM的从假基因区域下游至恒定区μ1的区域插入用于确认基因转变的序列(GC确认序列;由来源于人Igγ的可变区的序列构成的人假基因)、人IgG1可变区和人IgG1恒定区,制造图4所示的靶向载体。各个部分的制造方法如以下所示。(1)人IgG1恒定区使用上述1-6中记载的人IgG1恒定区序列。(2)人IgG1可变区使用上述1-6中记载的人IgG1可变区。(3)基因转变确认用序列基于上述1-6中设计的人Igγ的可变区,设计使CDR1缺失1个碱基、使CDR2缺失2个碱基、将CDR3插入1个碱基的变异序列,合成将其用禽Igγ假基因区域的非翻译区域序列连接的GC确认用序列(序列号42)。(4)左臂使用上述1-6中记载的左臂。(5)中臂1使用上述1-6中记载的中臂1。(6)中臂2使用上述1-6中记载的中臂2。(7)右臂使用上述1-6中记载的右臂。(8)靶向载体的构建在pBluescript中以成为图4表示的结构的方式组装上述的左臂、GC确认用序列、中臂1、人IgG1可变区、杀稻瘟菌素耐药性基因、中臂2、人IgG1恒定区、新霉素耐药性基因、右臂,得到humanHCKIvector。该载体的基因序列示于序列号43。1-8.表达Cre重组酶的载体的构建为了上述制造的载体的药剂耐药性基因以在两端添加loxP序列的形式组装在载体中,在基因导入后,通过使Cre重组酶工作能够除去该基因。因此,在pEGFP-C1(BDBioscienceCompany、#6084-1)中,组装序列号44表示的Cre重组酶基因,制造序列号45表示的Cre_EGFP-C1。2.将轻链插入·取代为人序列的细胞株的制造宿主使用来源于鸡的B细胞的细胞株DT40。DT40的培养方法利用以下方法进行。使用CO2恒温槽,在39.5℃、5%CO2存在下培养。培养基使用IMDM(LifeTechnologiesCorporation),添加9%FBS、1%鸡血清、青霉素100单位/mL、链霉素100μg/mL、单硫代甘油50μM而使用。以下,只要没有特别说明,“培养基”是指上述组成的培养基。为了评价轻链中的基因转变,准备下表所示的5种细胞株。[表1]制造方法如以下所示。2-1.将可变区·恒定区取代为人型的细胞株的制造利用限制性内切酶NotI将上述1-1中制备的humanLCKIvector制成直链状后,通过电穿孔法对DT40细胞进行基因导入。利用添加有2mg/mL的G418和10μg/mL的杀稻瘟菌素的培养基培养7~10天。对于繁殖的细胞进行利用PCR的基因分型,选择在目的基因座发生基因取代的细胞株。对该细胞株,基因导入上述1-8中制造的Cre_pEGFP-C1,通过利用流式细胞仪的单细胞分选选出GFP阳性细胞后,进行利用PCR的基因分型,得到能够确认药剂耐药性基因被除去的细胞株37-3。然后,利用限制性内切酶NotI将上述1-2中制造的KI-C-INvector制成直链状后,对细胞株37-3进行基因导入。利用添加有2mg/mL的G418的培养基培养7~10天。对繁殖的细胞进行利用PCR的基因分型,得到在目的基因座中产生基因插入的细胞株LP1(图5)。2-2.将可变区·恒定区取代为人型,进而在禽假基因区域的下游插入基因转变确认用序列(GC序列)的细胞株的制造利用限制性内切酶NotI将上述1-3中制备的KI-C-IN-Cvector制成直链状后,在上步得到的细胞株37-3中进行基因导入。利用添加有2mg/mL的G418的培养基培养7~10天。对繁殖的细胞进行利用PCR的基因分型,得到在目的基因座中产生基因插入的细胞株LP9(图6)。2-3.将可变区·恒定区取代为人型,进而使禽假基因区域缺失的细胞株的制造利用限制性内切酶NotI将上述1-4中制备的KI-C-DEvector制成直链状后,对上述2-1中得到的细胞株37-3进行基因导入。利用添加有2mg/mL的G418的培养基培养7~10天。对繁殖的细胞进行利用PCR的基因分型,得到在目的基因座中产生基因插入的细胞株LP18(图7)。2-4.将可变区·恒定区取代为人型,进而将禽假基因区域取代为基因转变确认用序列(GC序列)的细胞株的制造利用限制性内切酶NotI将上述1-5中制备的KI-C-DE-Cvector制成直链状后,在上述2-1中得到的细胞株37-3中进行基因导入。利用添加有2mg/mL的G418的培养基培养7~10天。对繁殖的细胞进行利用PCR的基因分型,得到在目的基因座中发生基因取代的细胞株LP20(图8)。3.抗体表达的确认(流式细胞仪、ELISA)3-1.利用流式细胞仪的解析进行在上述2中制造的细胞株的细胞表面表达的抗体的解析。培养各个细胞株,在96孔板(NuncCompany,249662)每孔加入5×105个,利用150μL的FACS缓冲液(含有0.3%牛血清白蛋白(BSA)的PBS)清洗2次后,添加将Goatanti-ChickenIgM-FITCconjugate(BethylCompany、A30-102F-20)、GoatAnti-HumanIgG(Gammachainspecific)R-PEconjugate(SouthernbiotechCompany,A2040-09)或GoatAnti-humanlambdaPEconjugate(SouthernbiotechCompany,2070-09)利用FACS缓冲液分别稀释至1000倍、500倍、500倍的1次抗体液50μL。在冰浴暗处孵育30分钟后,利用150μL的FACS缓冲液清洗2次,添加利用FACS缓冲液稀释1000倍的PI溶液200μL,混悬。将该混悬液提供于流式细胞仪解析,解析禽IgM和人Igγ(重链)或人Igλ(轻链)的表达,结果如图9所示,确认野生型DT40中仅禽IgM表达,而转化株LP1、LP9、LP18、LP20中禽IgM和人Igλ表达。在任何的细胞株中均没有发现未进行基因导入的Igγ的表达。3-2.利用ELISA的解析进行由上述2中制造的细胞分泌至培养上清中的禽IgM和人IgG1的浓度的测定。将各个细胞株利用不含有鸡血清的培养基混悬为4×105cells/mL之后,在24孔板每孔2mL进行播种,培养3天后,回收培养液。利用0.22μm过滤器过滤后,用于测定。IgG的测定方法如以下所示。将1.0μg/mL的Goatanti-HumanIgG-Fcaffinitypurified(BethylCompany,A80-104A)20μL分注至免疫384孔板Maxisorp(NuncCompany,464718),室温下反应1小时以上,在板上固相化。然后,利用清洗液(含有0.05%Tween20的PBS)清洗5次,添加封闭液(含有1%BSA的PBS)50μL,室温下反应30分钟。利用清洗液清洗5次,添加20μL测定样品或成为标准物质的HumanIgG1Lambda-UNLB(SouthernBiotechCompany,0151L-01),室温下反应1小时。利用清洗液清洗5次,添加利用含有1%BSA和0.05%Tween20的PBS将Goatanti-HumanIgG-FcHRPconjugated(BethylCompany,A80-104P)稀释1000倍的稀释液20μL,室温下反应1小时。利用清洗液清洗5次,添加TMB+(DakoCompany,S159985)20μL,室温下进行3分钟显色反应,添加1N硫酸20μL使反应停止。使用InfiniteM1000(TECANCompany),测定450nm的吸光度。IgM的测定中使用alphaLISAImmunoAssay(PerkinElmerCompany)的系统。对Goatanti-chickenIgMantibody(BethylCompany,A30-102A)使用ChromalinkTMBiotinLabelingReagent(SoluLinkInc.,#B1001-105)进行生物素标记,制备生物素化抗体。接下来,使Goatanti-chickenIgMantibody(BethylCompany,A30-102A)与UnconjugatedAlphaLISAAcceptorBeads(PerkinElmerCompany)结合,制备抗体结合AlphaScreenBeads。将该生物素标记抗体和抗体结合AlphaScreenBeads,利用检测缓冲液(含有1%BSA的PBS)分别稀释为4.5nM和50μg/mL之后,混合等量,作为禽IgM浓度测定溶液。96孔板(NuncCompany)中,混合禽IgM浓度测定溶液20μL和成为测定样品或标准物质的禽血清(GIBCOCompany、16110)5μL后,将每孔12.5μL分注至AlphaPlate-384(PerkinElmerCompany)。室温下反应60分钟后,添加利用检测缓冲液制备成80μg/mL的AlphaScreenstreptaVidinDonorBeads(PerkinElmerCompany)12.5μL,室温遮光反应30分钟。然后,使用EnSpire(PerkinElmerCompany)进行解析。测定结果示于表2。确认与野生型DT40同样,转化株LP1、LP9、LP18、LP20也分泌禽IgM。[表2]细胞株IgG(μg/mL)IgM(μg/mL)野生型<0.012.30LP1<0.010.09LP9<0.010.10LP18<0.010.09LP20<0.010.034.基因转变的确认将上述3中确认人Igλ(轻链)的表达的细胞株在含有2.5ng/mL的TSA的培养基中培养20天后,从1×106个细胞提取基因组DNA。将该基因组DNA作为模板,通过使用附有识别序列的正义引物(CGTATCGCCTCCCTCGCGCCATCAGNNNNNNNNNNCAGGTTCCCTGGTGCAGGC)(序列号46)和反义引物(CTATGCGCCTTGCCAGCCCGCTCAGGCTTGGTCCCTCCGCCGAA)(序列号47)的PCR扩增轻链可变区,利用琼脂糖凝胶提取进行精制后,使用QIAGENPCRPurificationKit(QIGENCompany)浓缩。使用RocheDiagnosticsCompany的第二代序列解析系统进行该样品的序列解析,通过与插入的人假基因序列相同的基因序列在可变区是否存在评价基因转变的有无。其结果示于表3。在不含有人假基因序列的LP1、LP18中确认不产生基因转变,而在具有人假基因序列的LP9、LP20中确认产生基因转变。[表3]细胞株序列数具有重组的序列数频率LP178500.0%LP9344185.2%LP1837800.0%LP20634314.9%5.将重链插入·取代为人序列的细胞株的制造为了评价重链中的基因转变,准备表4表示的3种细胞株。[表4]制造方法如以下所示。5-1.将重链的可变区·恒定区改变为人型的细胞株的制造利用限制性内切酶SalI将上述1-6中得到的humanHCV-Cvector制成直链状后,在上述2-2中得到的细胞株37-3中进行基因导入。利用添加有2mg/mL的G418和10μg/mL的杀稻瘟菌素的培养基培养7~10天。对繁殖的细胞进行利用PCR的基因分型,得到在目的基因座中产生基因的取代的细胞株T18(图10)。5-2.将重链的可变区·恒定区改变为人型,在假基因区域下游插入基因转变确认用序列(GC确认序列)的细胞株的制造利用限制性内切酶SalI将上述1-7中得到的humanHCKIvector制成直链状后,在上述2-2中得到的细胞株37-3中进行基因导入。利用添加有2mg/mL的G418和10μg/mL的杀稻瘟菌素的培养基培养7~10天。对繁殖的细胞进行利用PCR的基因分型,得到在目的基因座产生基因的取代的细胞株T11、T12(图11)。6.抗体表达的确认(流式细胞仪、ELISA)6-1.利用流式细胞仪的解析基于上述3-1记载的方法,确认上述5中制造的细胞中的抗体表达。其结果示于图12。确认野生型DT40中仅禽IgM表达,而转化株T18、T11、T12中禽IgM的表达消失,取而代之人Igγ和Igλ表达。6-2.利用ELISA的解析基于上述3-2记载的方法,进行由上述5制造的细胞分泌至培养上清中的抗体的浓度的测定。其结果示于表5。与6-1同样,野生型DT40中仅分泌禽IgM,而导入人IgG1的T18、T11、T12分泌人IgG1。[表5]细胞株IgG(μg/mL)IgM(μg/mL)野生型<0.012.30T181.000.03T110.40<0.01T120.69<0.017.基因转变的确认使用上述6中确认抗体表达的细胞株,进行基因转变的确认。将基于上述4记载的方法提取的基因组DNA作为模板,通过使用正义引物(CTATGCGCCTTGCCAGCCCGCTCAGTCCGTCAGCGCTCTCT)(序列号48)和附有识别Tag的反义引物(CGTATCGCCTCCCTCGCGCCATCAGNNNNNNNNNNCGGTGACCAGGGTGCCCTG)(序列号49)的PCR扩增重链可变区后,基于上述4记载的方法进行序列解析。其结果示于表6。确认在不合有人假基因的T18中完全不产生基因转变,而具有人假基因序列的T11、T12中产生基因转变。[表6]细胞株序列数具有重组的序列数频率T18112500.0%T11303841513.4%T1241083057.4%在轻链·重链各插入5个由来源于人可变区的序列构成的假基因区域而成的L5/H5细胞株的制造8.载体的制备8-1.对禽抗体(Igλ)假基因区域的敲入为了将禽Igλ的假基因区域取代为由来源于人Igλ的可变区的序列构成的假基因(人轻链假基因),制造图13所示的靶向载体。制造方法如以下所示。(1)人抗体轻链假基因序列(pVL5)的设计和制造基于不产生数据库上的人基因组的V(D)J重组的免疫球蛋白λ轻链可变区的基因序列,选择CDR1、CDR2、CDR3的序列各5个,通过组合1-1中克隆的Igλ可变区的框架序列来设计5个序列pVL001(序列号69)、pVL002(序列号70)、pVL003(序列号71)、pVL004(序列号72)和pVL005(序列号73),设计含有它们的人轻链假基因序列pVL5(序列号50),进行基因合成。(2)靶向载体的制造在上述1-4中制造的KI-C-DEvector的基础上,通过将GC确认用序列取代为上述的人抗体轻链假基因序列pVL5,制造humanLCpVKIvector。该载体的基因序列示于序列号51。8-2.禽抗体重链(IgM)恒定区的取代为了将禽IgM的重链恒定区取代为人IgG1,制造图14所示的靶向载体。制造方法如以下所示。(1)人IgG1恒定区使用上述1-6中制造的人IgG1恒定区。(2)左臂使用上述1-6中制造的中臂2。(3)右臂使用上述1-6中制造的右臂。(4)靶向载体的制造在pBluescript中组装上述的左臂、人IgG1恒定区序列、新霉素耐药性基因、右臂,制造humanCHKIvector。该载体的基因序列示于序列号52。8-3.对禽IgM可变·假基因区域的插入(1)人IgG1可变区使用上述1-6中制备的人IgG1可变区。(2)人抗体重链假基因序列(pVH5)的设计和制造基于数据库上的人免疫球蛋白重链可变区的序列,选择CDR1、CDR2、CDR3序列各5个,通过组合上述1-6中克隆的可变区的框架序列来设计5个序列pVH001(序列号89)、pVH002(序列号90)、pVH003(序列号91)、pVH004(序列号92)和pVH005(序列号93),设计含有它们的人抗体重链假基因序列pVH5(序列号53),进行基因合成。(3)左臂使用上述1-6中制备的左臂。(4)中臂使用上述1-6中制备的中臂1。(5)右臂使用上述1-6中制备的中臂2。被确认的右臂的序列示于序列号54。(6)靶向载体的制备在pBluescript中组装上述的左臂、人抗体重链假基因序列pVH5、中臂、人IgG1可变区序列、杀稻瘟菌素耐药性基因、右臂,得到humanpVHVHKIvector。该载体的基因序列示于序列号55。9.在禽抗体轻链和重链的基因座中导入各5个的人假基因的L5/H5细胞株的制造通过在上述2-2中得到的细胞株37-3中,基因导入上述8-2中制备的humanCHKIvector、上述8-3中制备的humanpVHVHKIvector、上述8-1中制备的humanLCpVKIvector,制造L5/H5细胞株细胞株(图15)。将利用限制性内切酶SalI制成直链状的humanCHKIvector基因导入37-3,利用含有2mg/mL的G418的培养基培养,对繁殖的细胞进行利用PCR的基因分型,确认在目的基因座产生取代。然后,基因导入Cre_pEGFP-C1,通过细胞分选将GFP阳性细胞选出后,进行利用PCR的基因分型,确认药剂耐药性基因被除去。接下来,基因导入利用限制性内切酶SacI制成直链状的humanpVHVHKIvector,利用含有10μg/mL的杀稻瘟菌素的培养基培养,对繁殖的细胞进行利用PCR的基因分型,确认在目的基因座产生取代。然后,基因导入Cre_pEGFP-C1,通过细胞分选将GFP阳性细胞选出后,进行利用PCR的基因分型,确认药剂耐药性基因被除去。最后,基因导入利用限制性内切酶NotI制成直链状的humanLCpVKIvector,利用含有2mg/mL的G418的培养基培养,对繁殖的细胞进行利用PCR的基因分型后,得到在目的基因座中产生取代的细胞株B-B10和J-D3(图16、图17)。10.抗体表达的确认(流式细胞仪、ELISA)10-1.利用流式细胞仪的解析基于上述3-1记载的方法,确认上述9中制造的细胞中的抗体表达。其结果示于图18。确认野生型DT40中仅禽IgM表达,而在转化株B-B10和J-D3中禽IgM的表达消失,取而代之人Igγ和Igλ表达。10-2.利用ELISA的解析基于上述3-2记载的方法,进行由上述9中制造的细胞分泌的抗体的测定。其结果示于表7。与10-1同样,确认野生型DT40中仅分泌禽IgM,而转化株B-B10和J-D3中分泌人IgG1。[表7]细胞株IgG(μg/mL)IgM(μg/mL)野生型<0.012.55B-B102.190.01J·D33.45<0.0111.基因转变的确认使用上述10中确认抗体表达的细胞株,进行基因转变的确认。将从基于上述4记载的方法从在TSA存在下培养42天的细胞提取的基因组DNA作为模板,通过使用附有识别序列的正义引物(CGTATCGCCTCCCTCGCGCCATCAGNNNNNNNNNNCAGGTTCCCTGGTGCAGGC)(序列号46)和反义引物(CTATGCGCCTTGCCAGCCCGCTCAGGCTTGGTCCCTCCGCCGAA)(序列号47)的PCR将轻链可变区扩增,通过使用正义引物(CTATGCGCCTTGCCAGCCCGCTCAGTCCGTCAGCGCTCTCT)(序列号56)和附有识别Tag的反义引物(CGTATCGCCTCCCTCGCGCCATCAGNNNNNNNNNNTGGGGGGGGTTCATATGAAG)(序列号57)的PCR将重链可变区扩增后,基于上述4记载的方法进行序列解析。B-B10中的轻链和重链的可变区的序列解析的结果分别示于表8和表9。在轻链和重链的任一个的可变区都确认含有插入的来源于人假基因的序列的克隆,确认通过基因转变产生多种序列。此外,在J-D3中也同样,确认通过基因转变产生多种序列。[表8]#%CDR1CDR2CDR310.183VLpVL003VL20.046不匹配VLpVL00330.046VLVLpVL004VL:在可变区插入的序列pVL001·005:在假基因区域插入的序列不匹配:与VL、pVL中任一个均不完全一致的序列(认为是部分的基因重组、突变所致)[表9]#%CDR1CDR2CDR3111.176pVH004VHVH20.131pVH001VHVH30.131pVH004不匹配VH40.065pVH003VHVHVH:在可变区插入的序列pVH001-005:在假基因区域插入的序列不匹配:与VL、pVL中任一个均不完全一致的序列(认为是部分的基因重组、突变所致)在禽抗体的轻链·重链基因座中各插入15个来源于人可变区的序列的L15/H15细胞株的制造12.载体的制备12-1.对禽抗体轻链(Igλ)可变·恒定区的敲入基于上述1-1记载的方法,制造图19表示的靶向用载体Lambda-VL-CLKIvector2.0。(1)人Igλ的可变区和恒定区使用上述1-1中制造的可变区和恒定区。(2)左臂使用上述1-1中制造的左臂。(3)中臂使用上述1-1中制造的中臂。(4)右臂基于上述1-1中确定的禽抗体轻链的恒定区的下游区域的序列,合成右臂。(5)靶向载体的构建在pBluescript中以成为图19表示的结构的方式装入上述的左臂、人Igλ可变区、中臂、人Igλ恒定区、右臂,得到Lambda-VL-CLKIvector2.0(序列号58)。12-2.禽抗体轻链(Igλ)假基因的敲出(1)人Igλ可变区使用上述1-1记载的可变区序列。(2)左臂使用上述1-2记载的左臂。(3)中臂使用上述1-1记载的左臂作为中臂。(4)右臂使用上述1-1记载的中臂作为右臂。(5)多克隆位点为了插入人抗体轻链假基因序列pVL15,设计序列号59表示的多克隆位点。(6)靶向载体的构建在pUC19中组装左臂、新霉素耐药性基因、多克隆位点、中臂、人Igλ可变区、杀稻瘟菌素耐药性基因、右臂,构建图19所示的靶向载体pVLKI_pUC19_step5_BrRev(序列号60)。12-3.对禽抗体轻链(Igλ)假基因·可变区的敲入(1)人抗体轻链假基因序列(pVL15)基于数据库上的人免疫球蛋白λ可变区的序列,选择15个CDR1、CDR2、CDR3序列,通过组合上述1-1中克隆的人Igλ可变区的框架序列来设计15个序列pVL101(序列号74)、pVL102(序列号75)、pVL103(序列号76)、pVL104(序列号77)、pVL105(序列号78)、pVL106(序列号79)、pVL107(序列号80)、pVL108(序列号81)、pVL109(序列号82)、pVL110(序列号83)、pVL111(序列号84)、pVL112(序列号85)、pVL113(序列号86)、pVL114(序列号87)、pVL115(序列号88),设计含有它们的人抗体轻链假基因序列pVL15(序列号61),进行基因合成。(2)靶向载体的构建在上述12-2中制造的pVLKI_pUC19_step6_BrRev的多克隆位点组装上述的人假基因序列pVL15,构建图19所示的靶向载体pVLKI_pUC19_step6_BrRev_pVL#odd(序列号62)。12-4.对禽抗体重链(IgM)恒定区的敲入使用上述8-2中制造的humanCHKIvector。12-5.对禽抗体重链(IgM)可变区的敲入(1)人IgG1可变区使用上述1-6记载的可变区序列。(2)左臂使用上述1-6记载的中臂。(3)右臂使用上述1-6记载的右臂。(4)靶向载体的构建在pUC19中组装左臂、人IgG1可变区、杀稻瘟菌素耐药性基因、右臂,制造图20所示的靶向载体H(X)0·hV(B)_074-009/No4(序列号63)。12-6.对禽抗体重链(IgM)假基因区域的敲入(1)人抗体重链假基因序列(pVH15)基于数据库上的人免疫球蛋白重链可变区的序列,选择15个CDR1、CDR2、CDR3,通过组合上述1-6中克隆的人IgG1可变区的框架序列来设计15个序列pVH101(序列号94)、pVH102(序列号95)、pVH103(序列号96)、pVH104(序列号97)、pVH105(序列号98)、pVH106(序列号99)、pVH107(序列号100)、pVH108(序列号101)、pVH109(序列号102)、pVH110(序列号103)、pVH111(序列号104)、pVH112(序列号105)、pVH113(序列号106)、pVH114(序列号107)、pVH115(序列号108),设计将它们连接的人抗体重链假基因序列pVH15(序列号64),进行基因合成。(2)左臂使用上述1-6记载的左臂。(3)右臂使用上述1-6记载的中臂。(4)多克隆位点为了插入人抗体重链假基因序列,设计序列号65表示的多克隆位点。(5)利用Cre重组酶的部位特异性重组(RMCE法)已知假基因序列区域如果变长,则利用同源重组的导入效率降低。因此,为了使用Cre重组酶能够简便插入假基因序列,决定利用RMCE法。因此,设计含有loxm3rev序列和loxm7revRE序列的RMCE用盒序列。(6)靶向载体的构建在pUC19中组装左臂、新霉素耐药性基因、RMCE用盒序列、多克隆位点、右臂、人IgG1可变区后,通过在多克隆位点组装人抗体重链假基因区域pVH15,制造如图20所示的靶向载体pVHKI_pUC19_step3_pVH#odd_15pVH_RMCE(序列号66)。13.细胞株的制造通过在DT40细胞中基因导入上述12-1中制备的Lambda-VL-CLKIvector2.0、上述12-2中制备的pVLKI_pUC19_step5_BrRev、上述12-3中制备的pVLKI_pUC19_step6_BrRev_pVL#odd、上述12-4中制备的humanCHKIvector、上述12-5中制备的H(X)0·hV(B)_074-009/No4和上述12-6中制备的pVHKI_pUC19_step3_pVH#odd_15pVH_RMCE,制造细胞株。首先,基因导入利用限制性内切酶SalI制成直链状的humanCHKIvector,利用含有2mg/mL的G418的培养基培养,对繁殖的细胞进行利用PCR的基因分型,确认在目的基因座中产生取代。然后,基因导入Cre_pEGFP-C1,通过细胞分选将GFP阳性细胞选出后,进行利用PCR的基因分型,确认药剂耐药性基因被除去。接下来,将利用限制性内切酶NotI制成直链状的Lambda-VL-CLKIvector2.0基因导入DT40,利用含有10μg/mL的杀稻瘟菌素的培养基培养,对繁殖的细胞进行利用PCR的基因分型,确认在目的基因座产生取代。然后,基因导入Cre_pEGFP-C1,通过细胞分选将GFP阳性细胞选出后,进行利用PCR的基因分型,确认药剂耐药性基因被除去。进一步,基因导入利用限制性内切酶NotI制成直链状的pVLKI_pUC19_step5_BrRev,利用含有2mg/mL的G418和10μg/mL的杀稻瘟菌素的培养基培养,对繁殖的细胞进行利用PCR的基因分型,确认在目的基因座产生取代。然后,基因导入Cre_pEGFP-C1,通过细胞分选将GFP阳性细胞选出后,进行利用PCR的基因分型,确认药剂耐药性基因被除去。接下来,基因导入利用限制性内切酶NotI制成直链状的pVLKI_pUC19_step6_BrRev_pVL#odd,利用含有2mg/mL的G418和10μg/mL的杀稻瘟菌素的培养基培养,对繁殖的细胞进行利用PCR的基因分型,确认在目的基因座产生取代。然后,基因导入Cre_pEGFP-C1,通过细胞分选将GFP阳性细胞选出后,进行利用PCR的基因分型,确认药剂耐药性基因被除去。最后,基因导入利用限制性内切酶ScaI制成直链状的H(X)0·hV(B)_074-009/No4和利用限制性内切酶NotI制成直链状的pVHKI_pUC19_step3_pVH#odd_15pVH_RMCE,利用含有2mg/mL的G418和10μg/mL的杀稻瘟菌素的培养基培养,对繁殖的细胞进行利用PCR的基因分型,确认在目的基因座产生取代。然后,基因导入Cre_pEGFP-C1,通过细胞分选将GFP阳性细胞选出后,进行利用PCR的基因分型,得到药剂耐药性基因被除去的细胞株A12-3、A12-4、A12-5、B7-3、B7-9和B7-11。14.抗体表达的确认(流式细胞仪、ELISA)14-1.利用流式细胞仪的解析基于上述3记载的方法,确认上述13制造的细胞中的抗体表达。其结果示于图21和图22。确认野生型DT40中仅禽IgM表达,而在转化株A12-3、A12-4、A12-5、B7-3、B7-9和B7-11中禽IgM的表达消失,取而代之人Igγ和Igλ表达(图21、22)。应予说明,由于图21中A12-5的Igλ的柱状图存在错误,因此添加了修正为优先权日前取得的正确数据的图22。14-2.利用ELISA的解析基于上述3-2记载的方法,进行由上述13制造的细胞分泌的抗体的测定。其结果示于表10。与10-1同样,确认野生型DT40中仅分泌禽IgM,而在转化株A12-3、A12-4、A12-5、B7-3、B7-9和B7-11中分泌人IgG1。[表10]细胞株IgG(μg/mL)IgM(μg/mL)野生型<0.012.54A12-30.52<0.01A12-40.51<0.01A12-50.650.01B7-30.620.01B7-90.760.02B7-110.770.0215.基因转变的确认使用上述10中确认抗体表达的细胞株,进行基因转变的确认。将基于上述4记载的方法从在TSA存在下培养21天或42天的细胞提取的基因组DNA作为模板,通过使用附有识别序列的正义引物(CGTATCGCCTCCCTCGCGCCATCAGNNNNNNNNNNCAGGTTCCCTGGTGCAGGC)(序列号46)和反义引物(CTATGCGCCTTGCCAGCCCGCTCAGGCTTGGTCCCTCCGCCGAA)(序列号47)的PCR扩增轻链可变区,通过使用正义引物(CTATGCGCCTTGCCAGCCCGCTCAGTCCGTCAGCGCTCTCT)(序列号56)和附有识别Tag的反义引物(CGTATCGCCTCCCTCGCGCCATCAGNNNNNNNNNNTGGGGGGGGTTCATATGAAG)(序列号57)的PCR扩增重链可变区后,基于在上述4记载的方法进行序列解析。其中,在TSA存在下培养21天的B7-11中的轻链和重链的可变区的序列解析结果分别示于表11和表12,在TSA存在下培养42天的B7-11中的轻链和重链的可变区的序列解析结果(上位10个)分别示于表13和表14。在轻链和重链的任一个的可变区中均确认含有来源于人假基因的序列这样的克隆,表明通过基因转变产生多种序列。此外,在其他细胞株中,同样也确认通过基因转变产生多种序列。[表11]#%CDR1CDR2CDR310.327VLpVL103VL20.327VLpVL102VL30.196VLVLpVL11440.196VLpVL105VL50.131VLpVL104VL60.131VLVLpVL11270.131pVL105VLVL80.065VL不匹配pVL10690.065VLpVL109VL100.065VLVLpVL110110.065VLpVL107VLVL:在可变区插入的序列pVL101-115:在假基因区域插入的序列不匹配:与VL、pVL中任一个均不完全一致的序列(认为是部分的基因重组、突变所致)[表12]#%CDR1CDR2CDR310.46pVH105VHVH20.345pVH108VHVH30.345VHpVH107不匹配40.345pVH107VHVH50.345pVH111pVH111VH60.46pVH112不匹配VH70.23pVH109VHVH80.23VHpVH112VH90.23pVH105不匹配VH100.115pVH108pVH108VH110.115VHVHpVH113120.115VHVHpVH109130.115pVH107VH不匹配140.115pVH103不匹配VH150.23pVH111不匹配VH160.23pVH111不匹配不匹配170.115pVH111VHVHVH:在可变区插入的序列pVH101·115:在假基因区域插入的序列不匹配:与VL、pVL中任一个均不完全一致的序列(认为利用部分的基因重组、突变引起的序列)[表13]#%CDR1CDR2CDR310.175VLpVL109VL20.175pVL105VLVL30.175pVL1.05pVL105VL40.116*VLpVL105VL50.116*VLpVL105VL60.116不匹配pVL105VL70.058VLpVL109不匹配80.058VLVLpVL11490.058VLpVL102VL100.058VLpVL108VLVL:在可变区插入的序列pVL:在假基因区域插入的序列不匹配:与VL和pVL均不完全一致的序列(认为是部分的基因重组、突变所致)*:CDR以外的部分的序列不同。[表14]#%CDR1CDR2CDR311.789pVH108VHVH21.022pVH112VHVH30.681pVH109VHVH40.596VHpVH112VH50.596pVH111不匹配VH60.511pVH107VHVH70.426pVH111pVH111VH80.341pVH111VHVH90.256pVH108pVH108VH100.256VHpVH107不匹配VH:在可变区插入的序列pVH:在假基因区域插入的序列不匹配:与VH和pVH均不完全一致的序列(认为是部分的基因重组、突变所致)在轻链中插入15个由来源于人可变区的序列构成的假基因区域、在重链中插入30个由来源于人可变区的序列构成的假基因区域而成的L15/H30细胞株的制造16.载体的制备16-1.对禽抗体轻链(Igλ)可变·恒定区的敲入使用上述12-1中制造的Lambda-VL-CLKIvector2.0。16-2.禽抗体轻链(Igλ)假基因的敲出使用上述12-2中制造的pVLKI_pUC19_step5_BrRev。16-3.对禽抗体轻链(Igλ)假基因·可变区的敲入使用上述12-3中制造的pVLKI_pUC19_step6_BrRev_pVL#odd。16-4.对禽抗体重链(IgM)恒定区的敲入使用上述8-2中制造的humanCHKIvector。16-5.禽抗体重链(IgM)可变区的敲入(1)人IgG1可变区使用上述1-6记载的可变区序列。(2)左臂使用上述1-6记载的左臂。(3)中臂使用上述1-6记载的中臂。(4)右臂使用上述1-6记载的右臂。(5)RMCE用盒序列使用上述12-6中设计的RMCE用盒序列。(6)靶向载体的构建在pUC19中组装左臂、新霉素耐药性基因、RMCE用盒序列、中臂、人IgG1可变区、右臂,制造图23所示的靶向载体pVHKI_pUC19_step5_BsrRev_RMCE_cassette(序列号109)。16-6.对禽抗体重链(IgM)假基因区域的敲入(1)人抗体重链假基因序列(pVH30)基于数据库上的人免疫球蛋白重链可变区的序列,选择30个CDR1、CDR2、CDR3序列,通过组合上述1-6中克隆的人IgG1可变区的框架序列来设计30个序列pVH101(序列号94)、pVH102(序列号95)、pVH103(序列号96)、pVH104(序列号97)、pVH105(序列号98)、pVH106(序列号99)、pVH107(序列号100)、pVH108(序列号101)、pVH109(序列号102)、pVH110(序列号103)、pVH111(序列号104)、pVH112(序列号105)、pVH113(序列号106)、pVH114(序列号107)、pVH115(序列号108)、pVH116(序列号110)、pVH117(序列号111)、pVH118(序列号112)、pVH119(序列号113)、pVH120(序列号114)、pVH121(序列号115)、pVH122(序列号116)、pVH123(序列号117)、pVH124(序列号118)、pVH125(序列号119)、pVH126(序列号120)、pVH127(序列号121)、pVH128(序列号122)、pVH129(序列号123)、pVH130(序列号124),设计将它们连接的人抗体重链假基因序列pVH30(序列号125),进行基因合成。(2)左臂使用上述1-6记载的左臂。(3)右臂使用上述1-6记载的中臂。(4)多克隆位点为了插入人抗体重链假基因序列,设计序列号65表示的多克隆位点。(5)靶向载体的构建在pUC19中组装左臂、新霉素耐药性基因、多克隆位点、右臂、人IgG1可变区后,通过在多克隆位点组装人抗体重链假基因区域pVH30,制造图24所示的靶向载体pVHKI_pUC19_step3_pVH#odd_even(序列号126)。17.细胞株的制造在DT40细胞中,通过基因导入上述12-1中制备的Lambda-VL-CLKIvector2.0、上述12-2中制备的pVLKI_pUC19_step5_BrRev、上述12-3中制备的pVLKI_pUC19_step6_BrRev_pVL#odd、上述12-4中制备的humanCHKIvector、上述16-5中制备的pVHKI_pUC19_step5_BsrRev_RMCE_cassette和上述16-6中制备的pVHKI_pUC19_step3_pVH#odd_even,制造细胞株。首先,基因导入利用限制性内切酶SalI制成直链状的humanCHKIvector,利用含有2mg/mL的G418的培养基培养,对繁殖的细胞进行利用PCR的基因分型,确认在目的基因座中产生取代。然后,基因导入Cre_pEGFP-C1,通过细胞分选将GFP阳性细胞选出后,进行利用PCR的基因分型,确认药剂耐药性基因被除去。接下来,将利用限制性内切酶NotI制成直链状的Lambda-VL-CLKIvector2.0基因导入DT40,利用含有10μg/mL的杀稻瘟菌素的培养基培养,对繁殖的细胞进行利用PCR的基因分型,确认在目的基因座产生取代。然后,基因导入Cre_pEGFP-C1,通过细胞分选将GFP阳性细胞选出后,进行利用PCR的基因分型,确认药剂耐药性基因被除去。进一步,基因导入利用限制性内切酶NotI制成直链状的pVLKI_pUC19_step5_BrRev,利用含有2mg/mL的G418和10μg/mL的杀稻瘟菌素的培养基培养,对繁殖的细胞进行利用PCR的基因分型,确认在目的基因座产生取代。然后,基因导入Cre_pEGFP-C1,通过细胞分选将GFP阳性细胞选出后,进行利用PCR的基因分型,确认药剂耐药性基因被除去。接下来,基因导入利用限制性内切酶NotI制成直链状的pVLKI_pUC19_step6_BrRev_pVL#odd,利用含有2mg/mL的G418和10μg/mL的杀稻瘟菌素的培养基培养,对繁殖的细胞进行利用PCR的基因分型,确认在目的基因座产生取代。然后,基因导入Cre_pEGFP-C1,通过细胞分选将GFP阳性细胞选出后,进行利用PCR的基因分型,确认药剂耐药性基因被除去。进一步,基因导入利用限制性内切酶NotI制成直链状的pVHKI_pUC19_step5_BsrRev_RMCE_cassette,利用含有2mg/mL的G418和10μg/mL的杀稻瘟菌素的培养基培养,对繁殖的细胞进行利用PCR的基因分型,确认在目的基因座产生取代。然后,基因导入Cre_pEGFP-C1,通过细胞分选将GFP阳性细胞选出后,进行利用PCR的基因分型,确认药剂耐药性基因被除去。最后,基因导入利用限制性内切酶NotI制成直链状的pVHKI_pUC19_step3_pVH#odd_even,利用含有2mg/mL的G418的培养基培养,对繁殖的细胞进行利用PCR的基因分型,确认在目的基因座产生取代。然后,基因导入Cre_pEGFP-C1,通过细胞分选将GFP阳性细胞选出后,进行利用PCR的基因分型,得到药剂耐药性基因被除去的细胞株#7-7、#11-3和#11-6。18.抗体表达的确认(流式细胞仪、ELISA)18-1.利用流式细胞仪的解析基于上述3记载的方法,确认上述16中制造的细胞中的抗体表达。其结果示于图25。确认在野生型DT40中仅禽IgM表达,而在转化株#7-7、#11-3和#11-6中禽IgM的表达消失,取而代之人Igγ和Igλ表达(图25)。18-2.利用ELISA的解析进行由上述17中制造的细胞分泌的抗体的测定。人IgG的测定基于上述3-2记载的方法进行。禽IgM的测定法如以下所示。将1.0μg/mL的Goatanti-chickenIgM(BethylCompany,A30-102A)20μL分注至免疫384孔板Maxisorp(NuncCompany,464718),室温下反应1小时以上,在板上固相化。然后,利用清洗液(含有0.05%Tween20的PBS)清洗5次,添加封闭液(含有1%BSA的PBS)50μL,室温下反应30分钟。利用清洗液清洗5次,添加成为测定样品或标准物质的禽血清(GIBCOCompany,16110)20μL,室温下反应1小时。利用清洗液清洗5次,添加将Goatanti-chickenIgMHRPconjugated(BethylCompany,A30-102P)用含有1%BSA和0.05%Tween20的PBS稀释至5000倍的稀释液20μL,室温下反应1小时。利用清洗液清洗5次,添加TMB+(DakoCompany、S159985)20μL,室温下进行显色反应3分种。添加1N硫酸20μL使反应停止。使用InfiniteM1000(TECANCompany)测定450nm的吸光度。其结果示于表15。与13-1同样,确认野生型DT40中仅分泌禽IgM,而转化株#7-7、#11-3和#11-6中分泌人IgG1。[表15]人IgG1(μg/mL)禽IgM(μg/mL)野生型<0.012.33#7-70.13<0.01#11-30.24<0.01#11-60.16<0.0119.基因转变的确认使用上述10中确认抗体表达的细胞株,进行基因转变的确认。将基于上述4记载的方法由在TSA存在下培养42天的细胞株提取的基因组DNA作为模板,通过使用附有识别序列的正义引物(CGTATCGCCTCCCTCGCGCCATCAGNNNNNNNNNNCAGGTTCCCTGGTGCAGGC)(序列号46)和反义引物(CTATGCGCCTTGCCAGCCCGCTCAGGCTTGGTCCCTCCGCCGAA)(序列号47)的PCR扩增轻链可变区,通过使用正义引物(CTATGCGCCTTGCCAGCCCGCTCAGTCCGTCAGCGCTCTCT)(序列号56)和附有识别Tag的反义引物(CGTATCGCCTCCCTCGCGCCATCAGNNNNNNNNNNTGGGGGGGGTTCATATGAAG)(序列号57)的PCR扩增重链可变区后,基于上述4记载的方法进行序列解析。其中,#11-6中的轻链和重链的可变区的序列解析结果(上位10个)分别示于表16和表17。在轻链和重链的任一个的可变区均确认含有追加插入的来源于人假基因的序列的克隆,表明产生比L15/H15细胞株进一步多种的序列。此外,在其他细胞株中也同样,确认通过基因转变产生多种序列。[表16]#%CDR1CDR2CDR310.166VLpVL105VL20.166pVL105VLVL30.125pVL113pVL113pVL11340.125VLpVL109VL50.083不匹配pVL105VL60.083VLpVL104pVL10470.083VLpVL113VL80.083pVL114pVL114pVL11490.042pVL109pVL109pVL109100.042pVL115pVL115pVL115VL:在可变区插入的序列pVL:在假基因区域插入的序列不匹配:与VL和pVL均不完全一致的序列(认为是部分的基因重组、突变所致)[表17]#%CDR1CDR2CDR311.077pVH123VHVH20.824VHpVH129VH30.760pVH104不匹配VH40.507pVH123不匹配VH50.444pVH117不匹配VH60.380pVH129pVH129VH70.380pVH126pVH112VH80.380VHpVH111不匹配90.317pVH102VH不匹配100.253pVH112不匹配VHVH:在可变区插入的序列pVH:在假基因区域插入的序列不匹配:与VH和pVH均不完全一致的序列(认为是部分的基因重组、突变所致)在轻链中插入30个由来源于人可变区的序列构成的假基因区域、在重链中插入30个由来源于人可变区的序列构成的假基因区域而成的L30/H30细胞株的制造20.对禽抗体轻链(Igλ)假基因·可变区的敲入(1)人抗体轻链假基因序列(pVL30)基于数据库上的人免疫球蛋白λ可变区的序列,选择30个CDR1、CDR2、CDR3序列,通过组合上述1-1中克隆的人Igλ可变区的框架序列来设计30个序列pVL101(序列号74)、pVL102(序列号75)、pVL103(序列号76)、pVL104(序列号77)、pVL105(序列号78)、pVL106(序列号79)、pVL107(序列号80)、pVL108(序列号81)、pVL109(序列号82)、pVL110(序列号83)、pVL111(序列号84)、pVL112(序列号85)、pVL113(序列号86)、pVL114(序列号87)、pVL115(序列号88)、pVL116(序列号127)、pVL117(序列号128)、pVL118(序列号129)、pVL119(序列号130)、pVL120(序列号131)、pVL121(序列号132)、pVL122(序列号133)、pVL123(序列号134)、pVL124(序列号135)、pVL125(序列号136)、pVL126(序列号137)、pVL127(序列号138)、pVL128(序列号139)、pVL129(序列号140)、pVL130(序列号141),设计含有它们的人抗体轻链假基因序列pVL30(序列号142),进行基因合成。(2)靶向载体的构建在上述16-2的pVLKI_pUC19_step6_BrRev的多克隆位点组装上述的人假基因序列pVL30,构建图26所示的靶向载体pVLKI_pUC19_step6_BrRev_#odd+even(序列号143)。21.细胞株的制造在上述17中制造的L15/H30细胞株中,通过基因导入上述20中制备的pVLKI_pUC19_step6_BrRev_#odd+even,制造细胞株。基因导入利用限制性内切酶NotI制成直链状的pVLKI_pUC19_step6_BrRev_#odd+even,利用含有2mg/mL的G418和10μg/mL的杀稻瘟菌素的培养基培养,对繁殖的细胞进行利用PCR的基因分型,确认在目的基因座产生取代。然后,基因导入Cre_pEGFP-C1,通过细胞分选将GFP阳性细胞选出后,进行利用PCR的基因分型,得到药剂耐药性基因被除去的细胞株#7-7-48-5、#7-7-48-7和#7-7-48-10。22.抗体表达的确认(流式细胞仪、ELISA)22-1.利用流式细胞仪的解析基于上述3记载的方法,确认上述21中制造的细胞中的抗体表达。其结果示于图27。确认在野生型DT40中仅禽IgM表达,而在转化株#7-7-48-5、#7-7-48-7和#7-7-48-10中禽IgM的表达消失,取而代之人Igγ和Igλ表达(图27)。22-2.利用ELISA的解析基于上述18-2记载的方法,进行由上述21中制造的细胞分泌的抗体的测定。其结果示于表18。与13-1同样,确认在野生型DT40中仅分泌禽IgM,而在转化株#7-7-48-5、#7-7-48-7和#7-7-48-10中分泌人IgG1。[表18]人IgG1(μg/mL)禽IgM(μg/mL)野生型<0.012.33#7-7-48-50.26<0.01#7-7-48-70.16<0.01#7-7-48-100.13<0.0123.基因转变的确认使用上述22中确认抗体表达的细胞株,进行基因转变的确认。将基于上述4记载的方法由在TSA存在下培养42天的细胞株提取的基因组DNA作为模板,通过使用附有识别序列的正义引物(CGTATCGCCTCCCTCGCGCCATCAGNNNNNNNNNNCAGGTTCCCTGGTGCAGGC)(序列号46)和反义引物(CTATGCGCCTTGCCAGCCCGCTCAGGCTTGGTCCCTCCGCCGAA)(序列号47)的PCR扩增轻链可变区,通过使用正义引物(CTATGCGCCTTGCCAGCCCGCTCAGTCCGTCAGCGCTCTCT)(序列号56)和附有识别Tag的反义引物(CGTATCGCCTCCCTCGCGCCATCAGNNNNNNNNNNTGGGGGGGGTTCATATGAAG)(序列号57)的PCR扩增重链可变区后,基于上述4记载的方法进行序列解析。其中,7-7-48-10中的轻链和重链的可变区的序列解析结果(上位10个)分别示于表19和表20。在轻链和重链的任一个的可变区中均确认含有追加插入的来源于人假基因的序列的克隆,表明通过增加假基因的个数产生更多种序列。此外,在其他细胞株中也同样,确认通过基因转变产生多种序列。[表19]#%CDR1CDR2CDR310.347*VLpVL118VL20.270pVL119VLVL30.154VLpVL123pVL11840.116pVL130pVL130pVL13050.116VLVLpVL12760.077pVL116pVL116pVL11670.077pVL119pVL119VL80.077VLpVL129pVL12990.077VLpVL105VL100.077*VLpVL118VLVL:在可变区插入的序列pVL:在假基因区域插入的序列不匹配:与VL和pVL均不完全一致的序列(认为是部分的基因重组、突变所致)*:CDR以外的部分的序列不同。[表20]#%CDR1CDR2CDR3133.191*pVH104不匹配VH28.085pVH129不匹配VH36.241pVH108不匹配VH45.106pVH111不匹配VH53.688**pVH126不匹配VH61.418pVH126pVH126VH71.418pVH101不匹配VH81.277*pVH104不匹配VH91.277**pVH126不匹配VH100.993*pVH104不匹配VHVH:在可变区插入的序列pVH:在假基因区域插入的序列不匹配:与VH和pVH均不完全一致的序列(认为是部分的基因重组、突变所致)*:CDR2的序列不同。**:CDR2的序列不同。在轻链中插入30个由来源于人可变区的序列构成的假基因区域、在重链中插入15个由来源于人可变区的序列构成的假基因区域的L30/H15细胞株的制造24.细胞株的制造在上述17中制造的L15/H15细胞株A12-4中,通过基因导入上述20中制备的pVLKI_pUC19_step6_BrRev_pVL#odd+even,制造细胞株。基因导入利用限制性内切酶NotI制成直链状的pVLKI_pUC19_step6_BrRev_pVL#odd+even,利用含有2mg/mL的G418和10μg/mL的杀稻瘟菌素的培养基培养,对繁殖的细胞进行利用PCR的基因分型,确认在目的基因座产生取代。然后,基因导入Cre_pEGFP-C1,通过细胞分选将GFP阳性细胞选出后,进行利用PCR的基因分型,得到药剂耐药性基因被除去的细胞株A77-3和A77-2。25.抗体表达的确认(流式细胞仪、ELISA)25-1.利用流式细胞仪的解析基于上述3记载的方法,确认上述24中制造的细胞中的抗体表达。其结果示于图28。确认在野生型DT40中仅禽IgM表达,而在转化株A77-3和A77-2中禽IgM的表达消失,取而代之人Igγ和Igλ表达(图28)。25-2.利用ELISA的解析基于上述3-2记载的方法,进行由上述24中制造的细胞分泌的抗体的测定。其结果示于表21。与13-1同样,确认在野生型DT40中仅分泌禽IgM,而在转化株A77-3和A77-2中分泌人IgG1。[表21]人IgG1(μg/mL)禽IgM(μg/mL)野生型<0.012.33A77-30.43<0.01A77-20.48<0.01利用RMCE法在轻链中插入30个由来源于人可变区的序列构成的假基因区域、在重链插入30个由来源于人可变区的序列构成的假基因区域的L30/H15f15f和L30/H15r15f细胞株的制造26.载体的制造26-1.对禽抗体重链(IgM)假基因区域,进一步插入15个正向构建的假基因(1)人抗体重链假基因序列(pVH15a)制造将上述16中设计的pVH116、pVH117、pVH118、pVH119、pVH120、pVH121、pVH122、pVH123、pVH124、pVH125、pVH126、pVH127、pVH128、pVH129、pVH130以正向连接的pVH15a(序列号144)。(2)载体的构建在pUC57-Amp载体中,插入loxm3rev序列、新霉素耐药性基因、SV40earlypolyAterminator、SV40polyadenylationregion、loxPforRE序列、pVH15a(正向)、loxm7revLE序列,制造图29所示的载体RMCE_pVH1stKI_pVH15evenFor(序列号145)。26-2.对禽抗体重链(IgM)假基因区域进一步插入15个反向构建的假基因在pUC57-Amp载体中,插入loxm3rev序列、新霉素耐药性基因、SV40earlypolyAterminator、SV40polyadenylationregion、loxPforRE序列、pVH15a(反向)、loxm7revLE序列,制造图29所示的载体RMCE_pVH1stKI_pVH15evenRev(序列号146)。27.细胞株的制造L30/H15f15f细胞株是在上述24中制造的L30/H15细胞株中,导入上述26-1中制备的RMCE_pVH1stKI_pVH15evenFor而制造的。将RMCE_pVH1stKI_pVH15evenFor与Cre_pEGFP-C1一同基因导入来诱导部位特异性重组后,利用含有2mg/mL的G418的培养基培养,对繁殖的细胞进行利用PCR的基因分型,得到确认在目的基因座中产生取代的细胞株1-6-1和1-7-3。L30/H15r15f细胞株是在上述24中制造的L30/H15细胞株中导入上述26-2中制备的RMCE_pVH1stKI_pVH15evenRev而制造的。将RMCE_pVH1stKI_pVH15evenRev与Cre_pEGFP-C1一同基因导入来诱导部位特异性重组后,利用含有2mg/mL的G418的培养基培养,对繁殖的细胞进行利用PCR的基因分型,得到确认在目的基因座中产生取代的细胞株2-1-1和7-3-2。28.抗体表达的确认(流式细胞仪、ELISA)28-1.利用流式细胞仪的解析基于上述3记载的方法,确认上述27中制造的细胞中的抗体表达。其结果示于图30。确认在野生型DT40中仅禽IgM表达,而在转化株1-6-1、1-7-3、2-1-1和7-3-2中禽IgM的表达消失,取而代之人Igγ和Igλ表达(图30)。28-2.利用ELISA的解析基于上述3-2记载的方法,进行由上述27中制造的细胞分泌的抗体的测定。其结果示于表22。与13-1同样,确认野生型DT40中仅分泌禽IgM,而在转化株1-6-1、1-7-3、2-1-1和7-3-2中分泌人IgG1。[表22]人IgG1(μg/mL)禽IgM(μg/mL)野生型<0.012.331-6-10.18<0.011-7-30.07<0.012-1-10.12<0.017-3-20.18<0.0129.基因转变的确认使用上述10中确认抗体表达的细胞株,进行基因转变的确认。将基于上述4记载的方法从在TSA存在下培养42天的细胞株提取的基因组DNA作为模板,通过使用附有识别序列的正义引物(CGTATCGCCTCCCTCGCGCCATCAGNNNNNNNNNNCAGGTTCCCTGGTGCAGGC)(序列号46)和反义引物(CTATGCGCCTTGCCAGCCCGCTCAGGCTTGGTCCCTCCGCCGAA)(序列号47)的PCR扩增轻链可变区,通过使用正义引物(CTATGCGCCTTGCCAGCCCGCTCAGTCCGTCAGCGCTCTCT)(序列号56)和附有识别Tag的反义引物(CGTATCGCCTCCCTCGCGCCATCAGNNNNNNNNNNTGGGGGGGGTTCATATGAAG)(序列号57)的PCR扩增重链可变区后,基于在上述4记载的方法进行序列解析。其中,1-6-1中的轻链和重链的可变区的序列解析结果(上位10个)分别示于表23和表24,7-3-2中的轻链和重链的可变区的序列解析结果(上位10个)分别示于表25和表26。轻链和重链的任一个的可变区均确认含有追加插入的来源于人假基因的序列的克隆,表明与L30/H30细胞株同样产生多种序列。此外,在其他细胞株中也同样确认通过基因转变产生多种序列。[表23]#%CDR1CDR2CDR310.935VLpVL102VL20.468VLpVL118VL30.271不匹配pVL102VL40.271*pVL119VLVL50.222VLVLpVL12960.172*pVL119VLVL70.172VLpVL105VL80.123VLpVL130VL90.098不匹配pVL122VL100.098VLpVL109VLVL:在可变区插入的序列pVL:在假基因区域插入的序列不匹配:与VL和pVL均不完全一致的序列(认为是部分的基因重组、突变所致)*:CDR以外的部分的序列不同。[表24]#%CDR1CDR2CDR316.393*pVH112不匹配VH20.586pVH109不匹配VH30.479pVH108不匹配VH40.479pVH117不匹配VH50.426pVH107不匹配VH60.320pVH124不匹配VH70.266*pVH112不匹配VH80.266VH不匹配pVH10990.160不匹配pVH125VH100.160*pVH112不匹配VHVH:在可变区插入的序列pVH:在假基因区域插入的序列不匹配:与VH和pVH均不完全一致的序列(认为是部分的基因重组、突变所致)*:CDR2的序列不同。[表25]#%CDR1CDR2CDR310.293VLpVL109VL20.267pVL119VLVL30.187*pVL105VLVL40.160VLVLpVL12950.133VLpVL101VL60.133VLpVL118VL70.133VLVLpVL12780.107pVL119pVL119VL90.080VLVLpVL123100.080*pVL105VLVLVL:在可变区插入的序列pVL:在假基因区域插入的序列不匹配:与VL和pVL均不完全一致的序列(认为是部分的基因重组、突变所致)*:CDR以外的部分的序列不同。[表26]#%CDR1CDR2CDR310.867*VH不匹配pVH10920.819*VH不匹配pVH10930.819VH不匹配pVH11640.337pVH1.09不匹配VH50.241*VH不匹配pVH10960.193pVH105不匹配VH70.193pVH117不匹配VH80.145pVH112不匹配VH90.145pVH126不匹配pVH116100.096pVH107不匹配pVH109VH:在可变区插入的序列pVH:在假基因区域插入的序列不匹配:与VH和pVH均不完全一致的序列(认为是部分的基因重组、突变所致)*:CDR2的序列不同。利用RMCE法在轻链插入30个由来源于人可变区的序列构成的假基因区域、在重链插入45个由来源于人可变区的序列构成的假基因区域的L30/H15f15f15f和L30/H15r15r15f细胞株的制造30.载体的制造30-1.对禽抗体重链(IgM)假基因区域进一步插入15个正向构建的假基因(1)人抗体重链假基因序列(pVH15b)基于数据库上的人免疫球蛋白重链可变区的序列新选择15个CDR1、CDR2、CDR3序列,制造将pVH131(序列号147)、pVH132(序列号148)、pVH133(序列号149)、pVH134(序列号150)、pVH135(序列号151)、pVH136(序列号152)、pVH137(序列号153)、pVH138(序列号154)、pVH139(序列号155)、pVH140(序列号156)、pVH141(序列号157)、pVH142(序列号158)、pVH143(序列号159)、pVH144(序列号160)、pVH145(序列号161)以正向连接的pVH15b(序列号162)。(2)载体的构建在pUC57-Amp载体中,插入loxm3rev序列、杀稻瘟菌素耐药性基因、SV40earlypolyAterminator、SV40polyadenylationregion、loxm7revRE序列、pVH15b(正向)、loxPforLE序列,制造图31所示的载体pVH15B-ver3_RMCE2nd_For(序列号163)。30-2.对禽抗体重链(IgM)假基因区域进一步插入15个反向构建的假基因在pUC57-Amp载体中插入loxm3rev序列、杀稻瘟菌素耐药性基因、SV40earlypolyAterminator、SV40polyadenylationregion、loxm7revRE序列、pVH15b(反向)、loxPforLE序列,制造图31所示的载体pVH15B-ver3_RMCE2nd_Rev(序列号164)。31.细胞株的制造L30/H15f15f15f细胞株是在上述2中制造的L30/H15f15f细胞株中,导入上述30-1中制备的pVH15B-ver3_RMCE2nd_For而制造的。将pVH15B-ver3_RMCE2nd_For与Cre_pEGFP-C1一同基因导入而诱导部位特异性重组后,利用含有10μg/mL的杀稻瘟菌素的培养基培养,对繁殖的细胞进行利用PCR的基因分型,得到确认在目的基因座产生取代的细胞株2-1-3、2-1-11、1-n-1和2-n-3。L30/H15r15r15f细胞株是在上述27中制造的L30/H15r15f细胞株中导入上述27-2中制备的pVH15B-ver3_RMCE2nd_Rev而制造的。将pVH15B-ver3_RMCE2nd_Rev与Cre_pEGFP-C1一同基因导入来诱导部位特异性重组后,利用含有10μg/mL的杀稻瘟菌素的培养基培养,对繁殖的细胞进行利用PCR的基因分型,得到确认在目的基因座产生取代的细胞株4-1-5、4-1-7和3-1-11。32.抗体表达的确认(流式细胞仪、ELISA)32-1.利用流式细胞仪的解析基于上述3记载的方法,确认上述31中制造的细胞中的抗体表达。其结果示于图32和图33。确认在野生型DT40中仅禽IgM表达,而在转化株2-1-3、2-1-11、1-n-1、2-n-3(以上,图32)、4-1-5、4-1-7和3-1-11(以上,图33)中禽IgM的表达消失,取而代之人Igγ和Igλ表达。32-2.利用ELISA的解析基于上述3-2记载的方法,进行由上述31中制造的细胞分泌的抗体的测定。其结果示于表27。与13-1同样,确认在野生型DT40中仅分泌禽IgM,而在转化株2-1-3、2-1-11、1-n-1、2-n-3、4-1-5、4-1-7和3-1-11中分泌人IgG1。[表271人IgG1(μg/mL)禽IgM(μg/mL)野生型<0.012.332-1-30.36<0.012-1-110.16<0.011-n-10.18<0.012-n-30.16<0.014-1-50.37<0.014-1-70.94<0.013-1-111.31<0.0133.基因转变的确认使用上述10中确认抗体表达的细胞株,进行基因转变的确认。将基于上述4记载的方法从在TSA存在下培养42天的细胞株提取的基因组DNA作为模板,通过使用附有识别序列的正义引物(CGTATCGCCTCCCTCGCGCCATCAGNNNNNNNNNNCAGGTTCCCTGGTGCAGGC)(序列号46)和反义引物(CTATGCGCCTTGCCAGCCCGCTCAGGCTTGGTCCCTCCGCCGAA)(序列号47)的PCR扩增轻链可变区,通过使用正义引物(CTATGCGCCTTGCCAGCCCGCTCAGTCCGTCAGCGCTCTCT)(序列号56)和附有识别Tag的反义引物(CGTATCGCCTCCCTCGCGCCATCAGNNNNNNNNNNTGGGGGGGGTTCATATGAAG)(序列号57)的PCR扩增重链可变区后,基于上述4记载的方法进行序列解析。其中,2-1-3中的轻链和重链的可变区的序列解析结果(上位10个)分别示于表28和表29,3-1-11中的轻链和重链的可变区的序列解析结果(上位10个)分别示于表30和表31。在轻链和重链的任一个的可变区均确认含有追加插入的来源于人假基因的序列的克隆,表明通过基因转变生成多种序列。此外,在其他细胞株中也同样,确认通过基因转变产生多种序列。[表28]#%CDR1CDR2CDR310.882*pVL119VLVL20.428**VLpVL118VL30.241pVL119pVL119VL40.187**VLpVL118VL50.187**VLpVL118VL60.134VLpVL119VL70.134VLpVL109VL80.134pVL105VLVL90.107*pVL119VLVL100.080VLpVL105VLVL:在可变区插入的序列pVL:在假基因区域插入的序列不匹配:与VL和pVL均不完全一致的序列(认为是部分的基因重组、突变所致)*,**:CDR以外的部分的序列不同。[表29]#%CDR1CDR2CDR312.226pVH112不匹配VH21.272pVH109不匹配VH30.636VHpVH112VH40.477pVH144不匹配VH50.477pVH105不匹配VH60.318pVH145不匹配VH70.318pVH137不匹配VH80.318*pVH108不匹配VH90.318*pVH108不匹配VH100.318VH不匹配pVH116VH:在可变区插入的序列pVH:在假基因区域插入的序列不匹配:与VH和pVH均不完全一致的序列(认为是部分的基因重组、突变所致)*:CDR2的序列不同。[表30]#%CDR1CDR2CDR311.000pVL105VLVL20.788pVL119VLVL30.515*VLpVL118VL40.455*VLpVL118VL50.182VLpVL105VL60.152**pVL119pVL119VL70.152VLpVL109VL80.121VLpVL104VL90.091**pVL119pVL119VL100.091VLpVL107VLVL:在可变区插入的序列pVL:在假基因区域插入的序列不匹配:与VL和pVL均不完全一致的序列(认为是部分的基因重组、突变所致)*,**:CDR以外的部分的序列不同。[表31]#%CDR1CDR2CDR311.524pVH109不匹配VH21.220pVH11.2不匹配VH30.915pVH117不匹配VH40.915pVH142不匹配VH50.610pVH108不匹配VH60.610pVH138不匹配VH70.610pVH107不匹配VH80.457pVH139不匹配VH90.457pVH102不匹配VH100.457pVH142不匹配不匹配VH:在可变区插入的序列pVH:在假基因区域插入的序列不匹配:与VH和pVH均不完全一致的序列(认为是部分的基因重组、突变所致)利用RMCE法在轻链中插入30个由来源于人可变区的序列构成的假基因区域、在重链中插入60个由来源于人可变区的序列构成的假基因区域的L30/H15f15f15f15f和L30/H15r15r15r15f细胞株的制造34.载体的制造34-1.对禽抗体重链(IgM)假基因区域进一步插入15个正向构建的假基因(1)人抗体重链假基因序列(pVH15c)基于数据库上的人免疫球蛋白重链可变区的序列,新选择15个CDR1、CDR2、CDR3序列,制造将pVH146(序列号165)、pVH147(序列号166)、pVH148(序列号167)、pVH149(序列号168)、pVH150(序列号169)、pVH151(序列号170)、pVH152(序列号171)、pVH153(序列号172)、pVH154(序列号173)、pVH155(序列号174)、pVH156(序列号175)、pVH157(序列号176)、pVH158(序列号177)、pVH159(序列号178)、pVH160(序列号179)以正向连接的pVH15c(序列号180)。(2)载体的构建在pUC57-Amp载体中,插入loxm3rev序列、新霉素耐药性基因、SV40earlypolyAterminator、SV40polyadenylationregion、loxPforRE序列、pVH15c(正向)、loxm7revLE序列,制造图34所示的载体pVH15A_RMCE1st_Fw(序列号181)。34-2.对禽抗体重链(IgM)假基因区域进一步插入15个反向构建的假基因在pUC57-Amp载体中,插入loxm3rev序列、新霉素耐药性基因、SV40earlypolyAterminator、SV40polyadenylationregion、loxPforRE序列、pVH15c(反向)、loxm7revLE序列,制造图34所示的载体pVH15A_RMCE1st_Rv(序列号182)。35.细胞株的制造L30/H15f15f15f15f细胞株是在上述31中制造的L30/H15f15f15f细胞株中导入上述34-1中制备的pVH15A_RMCE1st_Fw而制造的。将pVH15A_RMCE1st_Fw与Cre_pEGFP-C1一同基因导入来诱导部位特异性重组后,利用含有2mg/mL的G418的培养基培养,对繁殖的细胞进行利用PCR的基因分型,得到确认在目的基因座产生取代的细胞株1n1-3,1n1-4,1n1-5。L30/H15r15r15r15f细胞株是在上述31中制造的L30/H15r15r15f细胞株中导入上述34-2中制备的pVH15A_RMCE1st_Rv而制造的。将pVH15A_RMCE1st_Rv与Cre_pEGFP-C1一同基因导入来诱导部位特异性重组后,利用含有2mg/mL的G418的培养基培养,对繁殖的细胞进行利用PCR的基因分型,得到确认在目的基因座产生取代的细胞株3111-1。36.抗体表达的确认(流式细胞仪、ELISA)36-1.利用流式细胞仪的解析基于上述3记载的方法,确认上述35中制造的细胞中的抗体表达。其结果示于图35。确认在野生型DT40中仅禽IgM表达,而在转化株1n1-3、1n1-4、1n1-5和3111-1中禽IgM的表达消失,取而代之人Igγ和Igλ表达(图35)。36-2.利用ELISA的解析基于上述3-2记载的方法,进行由上述35中制造的细胞分泌的抗体的测定。其结果示于表32。与13-1同样,确认在野生型DT40中仅分泌禽IgM,而在转化株1n1-3、1n1-4、1n1-5和3111-1中分泌人IgG1。[表32]人IgG1(μg/mL)禽IgM(μg/mL)野生型<0.012.331n1-30.32<0.011n1-40.34<0.011n1-50.22<0.013111-10.49<0.0137.基因转变的确认使用上述10中确认抗体表达的细胞株,进行基因转变的确认。将基于上述4记载的方法从在TSA存在下培养42天的细胞株提取的基因组DNA作为模板,通过使用附有识别序列的正义引物(CGTATCGCCTCCCTCGCGCCATCAGNNNNNNNNNNCAGGTTCCCTGGTGCAGGC)(序列号46)和反义引物(CTATGCGCCTTGCCAGCCCGCTCAGGCTTGGTCCCTCCGCCGAA)(序列号47)的PCR扩增轻链可变区,通过使用正义引物(CTATGCGCCTTGCCAGCCCGCTCAGTCCGTCAGCGCTCTCT)(序列号56)和附有识别Tag的反义引物(CGTATCGCCTCCCTCGCGCCATCAGNNNNNNNNNNTGGGGGGGGTTCATATGAAG)(序列号57)的PCR扩增重链可变区后,基于上述4记载的方法进行序列解析。其中,1n1-3中的轻链和重链的可变区的序列解析结果(上位10个)分别示于表33和表34,3111-1中轻链和重链的可变区的序列解析结果(上位10个)分别示于表35和表36。在轻链和重链的任一个的可变区中均确认含有追加插入的来源于人假基因的序列的克隆,表明产生更多种的序列。此外,在其他细胞株中也同样,确认通过基因转变产生多种序列。[表33]#%CDR1CDR2CDR311.217不匹配pVL105VL20.466*不匹配pVL118VL30.304pVL119VLVL40.223**pVL119pVL119VL50.223*不匹配pVL118VL60.162不匹配pVL109VL70.162pVL105VLVL80.142不匹配pVL119VL90.081不匹配VLpVL129100.061**pVL119pVL119VLVL:在可变区插入的序列pVL:在假基因区域插入的序列不匹配:与VL和pVL均不完全一致的序列(认为是部分的基因重组、突变所致)*:CDR2的序列不同。**:CDR以外的部分的序列不同。[表34]#%CDR1CDR2CDR3163.115*pVH105不匹配VH22.186pVH138pVH138VH30.929pVH124不匹配VH40.820pVH112不匹配VH50.765*pVH105不匹配VH60.656*pVH105不匹配VH70.601pVH155不匹配VH80.601*pVH105不匹配VH90.546*pVH105不匹配VH100.492pVH156不匹配VHVH:在可变区插入的序列pVH:在假基因区域插入的序列不匹配:与VH和pVH均不完全一致的序列(认为是部分的基因重组、突变所致)*:CDR2的序列不同。[表35]#%CDR1CDR2CDR310.142VLVLpVL12920.094VLVLpVL12830.071*pVL119VLVL40.047*pVL119VLVL50.024不匹配pVL107pVL10760.024pVL108pVL123pVL12370.024pVL108pVL108pVL12380.024*pVL119VLVL90.024VLVLpVL125100.024VLVLpVL130VL:在可变区插入的序列pVL:在假基因区域插入的序列不匹配:与VL和pVL均不完全一致的序列(认为是部分的基因重组、突变所致)*:CDR以外的部分的序列不同。[表36]#%CDR1CDR2CDR3151.762*pVH126不匹配VH223.429pVH108不匹配VH31.053pVH129不匹配VH40.545*pVH126不匹配VH50.436*pVH126不匹配VH60.291pVH104不匹配VH70.291*pVH126不匹配VH80.291**pVH126不匹配不匹配90.254**pVH126不匹配不匹配100.254*pVH126不匹配VHVH:在可变区插入的序列pVH:在假基因区域插入的序列不匹配:与VH和pVH均不完全一致的序列(认为是部分的基因重组、突变所致)*:CDR2的序列不同。*:CDR2,CDR3的序列不同。抗体选择138.抗原的制备作为抗原蛋白质,选择PlexinA4。已知PlexinA4是属于PlexinAtype的分子,作为控制感觉神经、交感神经的轴突延伸的因子。抗原使用在PlexinA4的Sema结构域和PSI(Plexin-Semaphorin-Integrin)结构域的C末端添加FLAGTag的抗原(氨基酸序列示于序列号68)。基于ACCESSIONNo.NM_020911和ACCESSIONNo.EAW83796,合成序列号67表示的碱基序列,插入至pCEP4(LifeTechnologiesCorporation,V044-50)的使EBNA-1基因和潮霉素耐药性基因缺失的表达载体,构建PlexinA4表达载体。使用PEI(聚乙烯亚胺)将得到的PlexinA4表达载体导入FreeStyle293F细胞(LifeTechnologiesCorporation,R790-07),使其表达。此时,另外地将EBNA-1基因的表达载体也同时导入。表达为3L规模,培养进行7天。利用FLAGTag精制培养上清中的目的蛋白质。在XK16/20柱(GEHealthcareCompany,28-9889-37)中填充anti-M2-agarose树脂(SigmaCompany,A2220)2.5mL,利用D-PBS(-)(和光纯药,045-29795)平衡化后,载入培养上清,利用D-PBS(-)清洗后利用含有0.1mg/mL的FLAG肽(SigmaCompany,F3290)的D-PBS(-)洗脱。利用280nm的吸光度监测,回收相当于峰的成分,通过SDS-PAGE和CBB染色确认回收的蛋白质的分子量,确认与目的蛋白质的分子量一致。使用超滤膜(AmiconCompany,UFC0801008)将约24mL的回收馏分浓缩成2mL,使用HiLoad16/600Superdex200pg(GEHealthcareCompany,28-9893-35)进行利用凝胶过滤层析的精制。利用D-PBS(-)将柱平衡化后,载入回收馏分,利用D-PBS(-)洗脱。利用280nm的吸光度监测,回收相当于峰的成分,通过SDS-PAGE和CBB染色确认回收的蛋白质的分子量。39.抗体的选择39-1.抗原磁珠的制造磁珠使用DynabeadsM-270CarboxylicAcid(LifeTechnologiesCorporation,14305D)和DynabeadsM-270Epoxy(LifeTechnologiesCorporation,14301),与抗原的结合按照说明书进行。此时,磁架使用MPC-S(LifeTechnologiesCorporation,DBA13346)。对于DynabeadsM-270CarboxylicAcid,将珠10μL利用20μL的25mMMES(pH5.0)清洗3次后,顺次添加50mg/mL的NHS溶液10μL和50mg/mL的EDC溶液10μL,室温下反应30分钟后,利用20μL的25mMMES(pH5.0)清洗2次。从该磁珠混悬液除去上清后,添加利用25mMMES(pH5.0)制备为0.6mg/mL的PlexinA4蛋白质溶液10μL,4℃下过夜,边旋转搅拌边反应。除去上清后,添加淬灭缓冲液(50mMTris-HCl)20μL,室温下边旋转搅拌边反应15分钟。对于DynabeadsM-270Epoxy,利用20μL的100mM磷酸钠缓冲溶液(Na-phosphatebuffer)将珠10μL清洗3次后,添加利用100mM磷酸钠缓冲溶液制备成0.45mg/ml的PlexinA4蛋白质溶液13.3μL和含有3M硫酸铵的100mM磷酸钠缓冲溶液6.7μL进行混悬,37℃过夜,边旋转搅拌边反应。2种珠在将上清除去后收集至选择缓冲液(含有1%BSA的PBS)1mL进行混悬,利用相同缓冲液清洗3次后,在200μL选择缓冲液中混悬,以用于选择。39-2.利用抗原磁珠的选择将L15/H15细胞株B7-3在含有2.5ng/mL的TSA的培养基中培养3周后,利用选择缓冲液10mL清洗约1×108个细胞,除去上清之后利用选择缓冲液1mL清洗,然后在950μL的选择缓冲液中混悬。在该细胞混悬液中,添加上述17-1中制备的抗原磁珠50μL,4℃下边通过旋转进行搅拌边进行反应30分钟。使用KingFisherml(ThermoCompany,5400050),利用0.5mL选择缓冲液清洗3次。然后将回收的细胞利用20mL的培养基混悬后,在9cm培养皿中播种,在CO2培养箱中培养7天。39-3.利用流式细胞仪的筛选回收上述39-2的细胞培养液,分取3×106个细胞至1.5mL管并通过离心除去上清,利用1.5mL的PBS清洗,除去上清。使用EZ-Link(注册商标)NHS-PEG4-BiotinylationKit(PIERCECompany,21455)制备生物素标记的PlexinA4蛋白质,添加1次染色液(2.5μg/mL的生物素标记PlexinA4蛋白质溶液)300μL,以4℃遮光静置60分钟。通过离心除去上清,利用500μL的PBS清洗2次后,添加利用缓冲液分别将GoatAnti-HumanIgGgammachainspecificR-FITCconjugated(SouthernbiotechCompany,A2040-02)和StreptavidinPEconjugated(eBioscienceCompany,12-4317-87)稀释1000倍或500倍而制备的2次染色液300μL,以4℃遮光静置60分钟。通过离心除去上清,利用500μLPBS清洗2次后,利用500μLFACS缓冲液(含有以PBS稀释1000倍的PI溶液(InvitrogenCompany,P3566))混悬。对该样品进行流式细胞仪解析。其结果示于图36。确认PlexinA4蛋白质和IgGγ的信号均高的P5区的细胞团。39-4.利用抗原固定ELISA法的筛选回收上述39-3的细胞培养上清,通过使用PlexinA4抗原的抗原固定ELISA法,进行与PlexinA4抗原反应的克隆的筛选。首先,将2.5μg/mLPlexinA4抗原蛋白质和阴性对照的胰蛋白酶抑制剂(SigmaCompany,T6522)、链霉亲和素(NacalaiTesqueCompany,32243)或卵清蛋白(SigmaCompany,A5503)的溶液20μL分注至免疫384孔板Maxisorp(NuncCompany,464718),4℃下反应过夜,在板上固相化。然后,利用清洗液(含有0.05%Tween20的PBS)清洗3次,添加封闭液(含有1%BSA的PBS)45μL,室温下反应1小时。利用清洗液清洗3次,添加测定样品25μL,室温下反应1小时。利用清洗液清洗3次,添加以PBS缓冲液将Goatanti-HumanIgG-FcHRP-conjugated(BethylCompany,A80-104P)稀释至2000倍的稀释液25μL,反应1小时。利用清洗液清洗3次,添加TMB+(DakoCompany,S159985)25μL,显色反应5分钟,添加1M硫酸25μL使反应停止。使用InfiniteM1000(TECANCompany),测定450nm的吸光度。其结果示于图37。表明大部分的克隆中产生与PlexinA4特异性反应的抗体。通过将上述39-3记载的流式细胞仪和上述39-4记载的抗原固定ELISA法组合的筛选,能够确定与PlexinA4特异性结合的阳性克隆。应予说明,除了B7-3以外,在使用L15/H30细胞株#11-6、L30/H30细胞株7-7-48-10的选择中,也能够确定与PlexinA4特异性结合的阳性克隆。40.抗PlexinA4抗体的表达确认40-1.利用ELISA的解析在上述39-5中确定的阳性克隆中,对于#62和#20,进行分泌至培养上清中的禽IgM和人IgG1的浓度的测定。将各个细胞株在不含有鸡血清的培养基以4×105cells/mL混悬后,每个孔1mL地播种到24孔板,培养3天后,回收培养液,利用0.22μm过滤器过滤后,用于测定。IgG的测定方法如以下所示。将利用PBS稀释制备成1.0μg/mL的Goatanti-HumanIgG-Fcaffinitypurified(BethylCompany,A80-104A)100μL分注到F96MaxisorpNuncImmunoplate(NuncCompany,464718),4℃下反应1小时以上,在板上固相化。然后,利用清洗液(含有0.05%Tween20的PBS)清洗5次,添加封闭液(含有1%BSA的PBS)200μL,室温下反应95分钟。利用清洗液清洗5次,添加测定样品或成为标准物质的HumanIgG1Lambda-UNLB(SouthernBiotechCompany,0151L-01)100μL,室温下反应1小时。利用清洗液清洗5次,添加利用PBS将Goatanti-HumanIgG-FcHRPconjugated(BethylCompany,A80-104P)稀释至1000倍的稀释液100μL,室温下反应1小时。利用清洗液清洗3次,添加TMB+(DakoCompany,S159985)100μL,室温下显色反应3分钟,添加1M硫酸20μL使反应停止。使用InfiniteM1000(TECANCompany),测定450nm的吸光度。IgM的测定方法如以下所示。将利用PBS稀释为1.0μg/mL的Goatanti-chickenIgM(BethylCompany,A30-102A)100μL分注到F96MaxisorpNuncImmunoplate(NuncCompany,439454),4℃下反应过夜,在板上固相化。然后,利用清洗液(含有0.05%Tween20的PBS)清洗5次,添加封闭液(含有1%BSA的PBS)200μL,室温下反应95分钟。利用清洗液清洗5次,添加测定样品或成为标准物质的禽血清(GIBCOCompany,16110)100μL,室温下反应1小时。利用清洗液清洗5次,添加利用含有1%BSA和0.05%Tween20的PBS将Goatanti-chickenIgMHRPconjugated(BethylCompany,A30-102P)稀释至5000倍的稀释液100μL,室温下反应1小时。利用清洗液清洗5次,添加TMB+(DakoCompany,S159985)100μL,室温下显色反应3分钟,添加1N硫酸100μL使反应停止。使用InfiniteM1000(TECANCompany),测定450nm的吸光度。测定结果示于表37。克隆#20和克隆#62中,确认分泌人IgG1,禽IgM的分泌极少。[表37]克隆名称IgG(μg/mL)IgM(μg/mL)#625.790.186#201.540.23140-2.利用蛋白质印迹法的解析通过蛋白质印迹法,进行由17-4中得到的产生抗原反应性抗体的阳性克隆#62和#20分泌的抗体的分析。将各个细胞株在不含有鸡血清的培养基中混悬为4×105cells/mL之后,每孔1mL地播种至24孔板,培养3天后,回收细胞株的培养液,利用0.22μm过滤器(MilliporeCompany,SLGVJ13SL)过滤。用于Igγ和Igλ的检测时,使用利用蛋白A柱精制的样品,用于IgM的检测时,使用精制前的样品。对于还原条件下的检测中使用的样品(还原样品),等量添加TRIS-SDS-β巯基乙醇样品处理液(CosmoBioCompany,423437)并混合,对于非还原条件下的检测中使用的样品(非还原样品),等量添加TRIS-SDS样品处理液(CosmoBioCompany,423420)并混合。然后,还原样品98℃反应3分钟,非还原样品37℃反应30分钟后,利用4~20%聚丙烯酰胺凝胶(CosmoBioCompany,414879)进行电泳,转印至尼龙膜后,利用封闭缓冲液(含有1%BSA的PBS)封闭,使1次抗体(GoatAnti-HumanIgG-FcFragmentAb-HRP(BethylCompany,A80-104P)、GoatAnti-HumanIgGLambda-HRP(SouthernBiotechCompany,2070-05)、GoatAnti-chickenIgM-HRP(BethylCompany,A30-102P)反应。利用含有0.1%Tween20的PBS清洗3次后,通过利用ECLplus(GEHealthcareCompany,RPN2132)的化学发光检测禽IgM、人Igγ(重链)、人Igλ(轻链)。其结果示于图38~图40。在利用抗Igγ抗体的蛋白质印迹中,还原状态下检测到55kDa的条带,非还原状态下检测到约160kDa的条带(图38)。在利用抗Igλ抗体的蛋白质印迹中,还原状态下检测到25kDa的条带,非还原状态下检测到约160kDa的条带(图39)。此外,在利用抗IgM抗体的蛋白质印迹中没有检测到信号(图40)。由此,确认得到的阳性克隆产生的抗体是全长的人抗体。抗体选择241.抗原的制备作为抗原蛋白质,选择Semaphorin3A(Sema3A)。已知Sema3A是属于Sema3家族的分子,是作为控制感觉神经、交感神经的轴突延伸的因子。抗原使用在Sema3A的N末端附有HisTag和人碱性磷酸酶(AP)的His-AP-hSema3A(序列号183)。表达该蛋白质的稳定表达株HEK293由横浜市立大学医学部分子药理神经生物学教室提供。表达为4L规模,培养进行4天。利用HisTag精制培养上清中的目的蛋白质。利用A液(20mM磷酸钠,150mMNaCl,20mM咪唑(pH7.5))将HisTrapEXCEL5mL(GEHealthcareCompany,17-3712-06)平衡化后,载入培养上清,利用A液清洗。然后,通过从A液至B液(20mM磷酸钠,150mMNaCl,500mM咪唑(pH7.5))以25柱容量线性置换的梯度进行洗脱。以280nm的吸光度监测,回收相当于峰的馏分,通过SDS-PAGE和CBB染色确认回收的蛋白质的分子量,确认与目的蛋白质的分子量一致。使用超滤膜(AmiconCompany,UFC0801008)将约80mL的回收馏分浓缩至1.5mL,使用HiLoad16/600Superdex200pg(GEHealthcareCompany,28-9893-35)进行利用凝胶过滤层析的精制。利用洗脱缓冲液(50mM磷酸钠,300mMNaCl,400mM精氨酸,pH7.0)将柱平衡化后,载入回收馏分,利用洗脱缓冲液洗脱。利用280nm的吸光度监测,回收相当于峰的馏分,通过SDS-PAGE和CBB染色确认回收的蛋白质的分子量。接下来,为了将洗脱液中的精氨酸除去,以D-PBS(-)进行第2次凝胶过滤层析。同样利用280nm的吸光度监测,回收相当于峰的馏分,通过SDS-PAGE和CBB染色确认回收的蛋白质的分子量。42.抗体的选择42-1.抗原磁珠的制造磁珠使用DynabeadsM-270CarboxylicAcid(LifeTechnologiesCorporation,14305D)、DynabeadsM-270Epoxy(LifeTechnologiesCorporation,14301)和DynabeadsHis-TagIsolation&Pulldown(LifeTechnologiesCorporation,10103D),与抗原的结合按照说明书进行。此时,磁架使用MPC-S(LifeTechnologiesCorporation,DBA13346)。对于DynabeadsM-270CarboxylicAcid,利用80μL的25mMMES(pH5.0)将珠40μL清洗3次后,顺次添加50mg/mL的NHS溶液40μL和50mg/mL的EDC溶液40μL,室温下反应30分钟后,利用80μL的25mMMES(pH5.0)清洗2次。从该磁珠混悬液除去上清后,正选择用时添加利用25mMMES(pH5.0)制备为0.475mg/ml的His-AP-hSema3A蛋白质溶液50.6μL并混悬,负选择用时代替抗原溶液添加使用PBS并以25mMMES(pH5.0)稀释的溶液50.6μL并混悬,4℃下边旋转搅拌边反应过夜。除去上清后,添加淬灭缓冲液(50mMTris-HCl)80μL,室温下边旋转搅拌边反应15分钟。对于DynabeadsM-270Epoxy,利用80μL的100mM磷酸钠缓冲溶液将珠40μL清洗3次后,正选择用时添加以100mM磷酸钠缓冲溶液制备为0.45mg/mL的His-AP-hSema3A蛋白质溶液53.3μL以及含有3M硫酸铵的100mM磷酸钠缓冲溶液26.7μL并混悬,负选择用时代替抗原溶液添加使用PBS并以25mMMES(pH5.0)稀释的溶液53.3μL以及含有3M硫酸铵的100mM磷酸钠缓冲溶液26.7μL并混悬,37℃下边旋转搅拌边反应过夜。对于DynabeadsHis-TagIsolation&Pulldown,利用80μL的PBS将珠8μL清洗3次后,正选择用时添加抗原浓度4.49μM的His-AP-hSema3A蛋白质溶液101.9μL并混悬,负选择用时代替抗原溶液添加PBS101.9μL并混悬,4℃下边旋转搅拌边反应10分钟。3种珠在除去上清后收集至选择缓冲液(含有1%BSA的PBS)1mL并混悬,利用相同缓冲液清洗3次后,在200μL的选择缓冲液中混悬,用于选择。42-2.利用抗原磁珠的选择将L15/H15细胞株B7-3在含有2.5ng/mL的TSA的培养基中培养86天后,将约1.5×107个细胞利用选择缓冲液10mL清洗,将上清除去后,利用选择缓冲液1mL清洗,然后在950μL的选择缓冲液中混悬。向该细胞混悬液中,添加上述39-1中制备的负选择用抗原磁珠50μL,4℃下边通过旋转进行搅拌边反应30分钟。使用KingFishermL(ThermoCompany,5400050)取出磁珠后,向细胞混悬液中添加正选择用抗原磁珠50μL,4℃下边旋转搅拌边反应30分钟。使用KingFishermL(ThermoCompany,5400050),利用1.7mL的选择缓冲液清洗3次。然后将回收的细胞利用20mL的培养基混悬后,播种到1张96孔板,利用CO2培养箱培养。42-3.1次筛选回收上述42-2的细胞培养液并基于抗原固定ELISA中的分泌IgG的抗原反应性,筛选与靶抗原特异性反应的克隆。将利用PBS制备成2.5μg/mL的抗原蛋白质和阴性对照抗原溶液(His-泛素,卵清蛋白,链霉亲和素)20μL分注到免疫384孔板Maxisorp(NuncCompany,464718),4℃下反应过夜进行固相化。利用清洗溶液(含有0.05%Tween20的PBS)清洗5次,添加封闭溶液(含有1%BSA的PBS)45μL室温下反应115分钟。利用清洗溶液清洗5次,添加培养上清25μL室温下反应130分钟。利用清洗溶液清洗5次,添加利用封闭溶液稀释至2000倍的Goatanti-HumanIgG-FcHRP-conjugated25μL在室温下反应47分钟。利用清洗溶液清洗5次,添加TMB+(DakoCompany,S159985)25μL反应5分钟。添加1N硫酸25μL使反应停止后,使用酶标仪测定450nm的吸光度(图41)。将吸光度0.1以上、S/N比5倍以上的细胞判定为阳性,用于2次筛选。42-4.2次筛选回收上述42-3的细胞培养上清,基于42-3记载的方法进行利用抗原固定ELISA的2次筛选。将吸光度0.5以上、S/N比5倍以上的细胞判定为阳性,进行单克隆化。42-5.单克隆化后的1次筛选回收上述42-4的细胞培养上清,基于42-3记载的方法进行利用抗原固定ELISA的筛选。将吸光度0.2以上、S/N比5倍以上的细胞判定为阳性,用于2次筛选。42-6.单克隆化后的2次筛选回收上述42-5的细胞培养上清,基于42-3记载的方法进行利用抗原固定ELISA的筛选。选择吸光度0.5以上、S/N比5倍以上的阳性克隆#64、#69和#77(图42)。43.抗His-AP-hSema3A抗体的表达确认43-1.利用ELISA的解析上述42-6中选择的阳性克隆中,对于#64,进行分泌到培养上清中的禽IgM和人IgG1的浓度的测定。测定法按照上述40-1进行。测定结果示于表38。与野生型DT40同样,确认由阳性克隆#64分泌人IgG,未发现禽IgM的分泌。[表38]克隆名称IgG(μg/mL)禽IgM(μg/mL)#642.66<0.000543-2.利用蛋白质印迹法的解析通过蛋白质印迹法,进行由产生上述42-6中得到的抗原反应性抗体的阳性克隆#64分泌的抗体的分析。将各个细胞株在不含有鸡血清的培养基中以4×105cells/mL混悬后,在9cm培养皿中播种20mL,培养4天后,回收细胞株的培养液,利用0.22μm过滤器(MilliporeCompany,SLGVJ13SL)过滤。Igγ和Igλ的检测用中使用利用蛋白A柱精制的样品,在IgM的检测用中使用精制前的样品。对于还原条件下的检测中使用的样品(还原样品),等量添加TRIS-SDS-β巯基乙醇样品处理液(CosmoBioCompany,423437)并混合,对于非还原条件下的检测中使用的样品(非还原样品),等量添加TRIS-SDS样品处理液(CosmoBio,423420)并混合。然后,还原样品98℃反应3分钟,非还原样品37℃之应30分钟后,利用XVPANTERA5-20%T-HCL10W(DRC、NXV-275HP)进行电泳,转印至PVDF膜后,利用封闭缓冲液(含有5%脱脂牛奶和0.1%Tween20的TBS)封闭,使1次抗体(GoatAnti-HumanIgG-FcFragmentAb-HRP(BethylCompany,A80-104P)、GoatAnti-HumanIgGLambda-HRP(SouthernBiotechCompany,2070-05)反应。利用含有0.1%Tween20的TBS清洗3次后,通过利用LuminataForteWesternHRPsubstrate(MilliporeCompany,WBLUF0100)的化学发光检测禽IgM、人Igγ(重链)、人Igλ(轻链)。其结果示于图43。利用抗Igγ抗体的蛋白质印迹中,还原状态下检测55kDa(图43A)的条带,非还原状态下检测约160kDa的条带(图43B)。利用抗Igλ抗体的蛋白质印迹中,还原状态下检测25kDa的条带(图43C),非还原状态下检测约160kDa的条带(图43D)。由此,确认得到的阳性克隆产生的抗体是全长的人抗体。抗体选择344.抗原的制备作为抗原蛋白质,选择IL-8。IL-8是趋化因子的一种,对嗜中性粒细胞、T淋巴细胞具有趋化性,具有促进白细胞与血管内皮细胞的粘合、嗜中性粒细胞的功能的活性。因此,认为参与伴随嗜中性粒细胞的浸润的炎症疾病、风湿性关节炎等。抗原在选择中使用人IL-8(ImmuneTECHCompany,IT-401-003P),在筛选中使用2种人IL-8(ImmuneTECHCompany,IT-401-003P和CELLSignalingTechnologyCompany,8921LF)。45.抗体的选择45-1.抗原磁珠的制造磁珠使用DynabeadsM-270CarboxylicAcid(LifeTechnologiesCorporation,14305D)、DynabeadsM-270Epoxy(LifeTechnologiesCorporation,14301)和DynabeadsHis-TagIsolation&Pulldown(LifeTechnologiesCorporation,10103D),与抗原的结合按照说明书进行。此时,磁架使用MPC-S(LifeTechnologiesCorporation,DBA13346)。对于DynabeadsM-270CarboxylicAcid,利用70μL的25mMMES(pH5.0)将珠35μL清洗3次后,顺次添加50mg/mL的NHS溶液35μL和50mg/mL的EDC溶液35μL,室温下反应30分钟后,利用70μL的25mMMES(pH5.0)清洗2次。从该磁珠混悬液除去上清后,在正选择用时添加利用25mMMES(pH5.0)制备为0.6mg/ml的IL-8(ImmuneTECHCompany,IT-401-003P)蛋白质溶液35μL并混悬,在负选择用时代替抗原溶液添加使用PBS并利用25mMMES(pH5.0)稀释的溶液35μL并混悬,4℃下边旋转搅拌边反应过夜。除去上清后,添加淬灭缓冲液(50mMTris-HCl)70μL,室温下边旋转搅拌边反应15分钟。对于DynabeadsM-270Epoxy,利用70μL的100mM磷酸钠缓冲溶液将珠35μL清洗3次后,在正选择用时添加利用100mM磷酸钠缓冲溶液制备为0.45mg/mL的IL-8蛋白质溶液46.7μL以及含有3M硫酸铵的100mM磷酸钠缓冲溶液23.3μL并混悬,在负选择用时代替抗原溶液添加使用PBS并利用100mM磷酸钠缓冲溶液稀释的溶液46.7μL以及含有3M硫酸铵的100mM磷酸钠缓冲溶液23.3μL并混悬,37℃下边旋转搅拌边反应过夜。对于DynabeadsHis-TagIsolation&Pulldown,利用70μL的PBS将珠7μL清洗3次后,在正选择用时添加抗原浓度5.75μM的IL-8蛋白质溶液70μL并混悬,在负选择用时代替抗原溶液添加PBS70μL并混悬,4℃下边旋转搅拌边反应210分钟。3种珠在除去上清后收集至选择缓冲液(含有1%BSA的PBS)1mL并混悬,利用相同缓冲液清洗3次后,在700μL的选择缓冲液中混悬,用于选择。45-2.利用抗原磁珠的选择将L30/H15f15f15f细胞株2-1-3在含有2.5ng/mL的TSA的培养基中培养45天后,利用选择缓冲液10mL将约1.5×107个的细胞清洗,除去上清后利用选择缓冲液10mL清洗,然后在950μL的选择缓冲液中混悬。在该细胞混悬液中,添加上述39-1中制备的负选择用抗原磁珠50μL,4℃下边通过旋转进行搅拌边反应30分钟。使用KingFishermL(ThermoCompany,5400050)将磁珠取出后,在细胞混悬液中添加正选择用抗原磁珠50μL,4℃下边旋转搅拌边反应30分钟。使用KingFisherml(ThermoCompany,5400050),利用1.7mL的选择缓冲液清洗3次。然后将回收的细胞利用20mL的培养基混悬后,播种至1张96孔板,利用CO2培养箱培养。45-3.筛选和阳性克隆的确定与上述42-3~42-6同样,实施2次筛选、单克隆化、2次筛选,得到阳性克隆#117、#121和#123。选择后的1次筛选的结果示于图44,单克隆化后的2次筛选的结果示于图45。46.抗IL-8抗体的表达确认46-1.利用ELISA的解析对于上述45-3中得到的产生抗原反应性抗体的阳性克隆#117和#121,进行由分泌至培养上清中的禽IgM和人IgG1的浓度的测定。测定法按照上述40-1进行。测定结果示于表39。与野生型DT40同样,确认由阳性克隆#117和#121分泌人IgG。未发现禽IgM的分泌。[表39]克隆名称IgG(μg/mL)IgM(μg/mL)#1173.65<0.041#1213.7<0.04146-2.利用蛋白质印迹法的解析通过蛋白质印迹法,进行由#117、#121分泌的抗体的分析。方法按照上述40-2。其结果示于图46。在利用抗Igγ抗体的蛋白质印迹中,还原状态下检测55kDa(图46A)的条带、非还原状态下检测约160kDa的条带(图46B)。在利用抗Igλ抗体的蛋白质印迹中,还原状态下检测25kDa的条带(图46C),非还原状态下检测约160kDa的条带(图46D)。由此,确认得到的阳性克隆产生的抗体是全长的人抗体。产业上的可利用性本发明涉及迅速制造多种人抗体的方法。鉴于抗体医药的重要性,在今后的药物研发、医疗领域,在开发具有期望的药效的生物医药品、特别是抗体医药时,期待本发明提供的技术发挥极其重要的作用。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1