用于恶性肿瘤的RNA干扰组合物和方法与流程
本发明涉及由基于核酸的分子构成的治疗剂和生物药物领域。具体而言,本发明涉及用于递送rna干扰剂的组合物和方法,所述rna干扰剂用于对恶性肿瘤的相关疾病和症状进行预防、治疗或疗效改善。序列表本申请包括以电子版形式提交的序列表ascii文件,该文件创建于2016年1月2日,文件名为nd5123179wo_sl.txt,大小为140,959字节,其全部内容通过参考并入本说明书中。
背景技术:
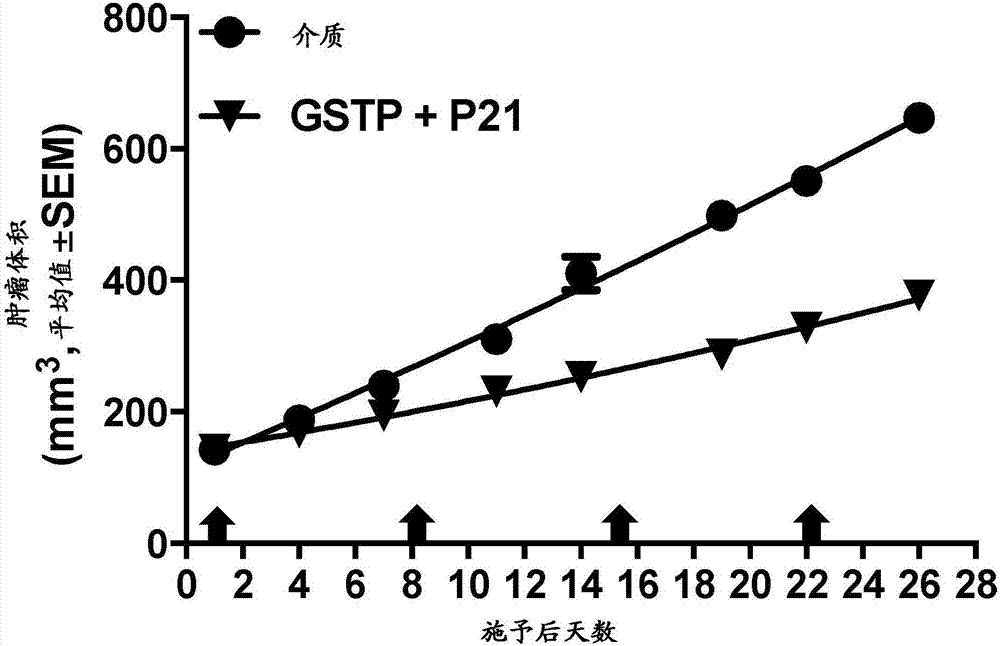
:kras基因突变可能与例如肺腺癌、粘液腺癌、胰腺导管癌、和结肠直肠癌等恶性肿瘤相关。最近的观察表明,该kras突变与谷胱甘肽s-转移酶-π(gst-π)蛋白水平的升高有关。在不拘泥于任何特定理论的前提下,已经发现,当细胞中的gst-π受到阻遏时,出乎意料地,细胞周期调控蛋白p21的水平显著升高。细胞周期调控蛋白p21的功能之一在于抑制细胞凋亡。例如,p21可能具有保护细胞、使其免于被化疗剂诱导凋亡的效果(体内和体外)。例如,参见gartelandtyner,2002,molcancerther.,2002,1(8):639-49;abbasanddutta,2009,natrevcancer.,2009,9(6):400-14。p21由cdkn1a基因编码,属于cip/kip家族。p21可通过与细胞周期蛋白-cdk复合物结合而发挥作用,在gl期和g2/m期抑制细胞周期的进程。例如,p21基因被肿瘤阻遏基因p53激活。当p53由于dna损伤而被激活时,其激活p21,从而使细胞周期停滞在gl期和g2/m期。gst-π是谷胱甘肽s-转移酶(iubmbec2.5.1.18)家族的6个同工酶之一,其通过催化疏水性和亲电子性的化合物与还原型谷胱甘肽缀合而参与解毒。作为多态性基因,gst-π基因(gstpl)编码多种高活性、功能各异的gstpl变体蛋白,这些蛋白被认为作用于异物代谢(xenobioticmetabolism)。gstpl还可能与癌症易感性有关,并在肿瘤细胞中大量表达。例如,参见aliyas.etal.molcellbiochem.,2003nov;253(1-2):319-327。在人体中,谷胱甘肽s-转移酶-π由gstpl基因编码。例如,参见boraps,etal.(oct1991)j.biol.chem.,266(25):16774-16777。gst-π同工酶还被发现催化gsh与某些烷基化抗癌剂的缀合,这暗示了gst-π的过量表达可能会导致肿瘤细胞的耐受性。在患有各种胃肠道恶性肿瘤(包括胃癌、食道癌、结肠癌、胰腺癌、肝细胞癌和胆管癌)的患者中,观察到gst-π的血清水平升高。80%以上患有iii期或iv期胃癌的患者、甚至约50%的ii期或ii期患者中,gst-π的血清水平升高。例如,参见niitsuy,etal.cancer,1989jan15;63(2):317-23。gst-π被发现作为用于预测口腔癌患者化疗后肿瘤复发的标志物有用。例如,参见hiratas.etal.cancer,1992nov15:70(10):2381-7。在人结肠直肠癌中,kras突变通过激活ap-1来诱导gst-π的过量表达。例如,参见miyanishietal.,gastroenterology,2001;121(4):865-74。gst-π的表达在各种癌细胞中升高,这可能与对于某些抗癌剂的耐受性有关。例如,参见banetal.,cancerres.,1996,56(15):3577-82;nakajimaetal.,jpharmacolexpther.,2003,306(3):861-9。已经公开了gst-π阻遏剂可诱导细胞凋亡。然而,这些组合物和技术也会导致自体吞噬,并且需要各种药剂的协同作用。例如,参见us2014/0315975al。另外,尚未发现阻遏gst-π会使肿瘤缩小或减轻。例如,在gst-π过量表达的癌症中,虽然观察到了其他的效果,但阻遏gst-π并未影响肿瘤重量。例如,参见hokaiwadoetal.,carcinogenesis,2008,29(6):1134-1138。当前,急需用于治疗恶性肿瘤患者的方法和组合物,例如可抑制gst-π和p21的表达的sirna序列、化合物和构建体。为了解决上述课题,需要用于预防或治疗恶性肿瘤的方法和组合物。并且,持续需求用于预防、治疗或减轻恶性肿瘤的rnai分子、其他构建体和组合物。技术实现要素:本发明提供涉及以人gst-π作为靶标的rnai分子、及以人p21作为靶标的rnai分子的组合物和方法。所述组合物还含有药学上允许的担体。本发明涉及用于恶性肿瘤的生物药物和治疗剂的分子和组合物。具体而言,本发明涉及用于提供下述纳米粒子的化合物、组合物和方法,所述纳米粒子将活性剂或药物化合物递送及分配至细胞、组织、器官和患有恶性肿瘤的受试者。本发明包括对有需要的受试者的恶性肿瘤的一种或多种症状进行预防、治疗或改善的方法。所述方法可包括向受试者施予有效量的以gst-π和p21作为靶标的rnai分子的组合物。本发明包括以下实施方式:组合物,其含有以人gst-π作为靶标的rnai分子、以人p21作为靶标的rnai分子、和药学上允许的担体。上述rnai分子可在反义链自3’端起第2~8位含有一个或多个2’-脱氧核苷酸。上述担体可包含包封rnai分子的脂质体纳米粒子。所述脂质体纳米粒子能够包封rnai分子,并且,在人血清中暴露1小时后,所述脂质体纳米粒子保持至少80%的包封的rnai分子。所述脂质体纳米粒子的大小可为10~1000nm或10~150nm。组合物具有恶性肿瘤治疗活性,所述恶性肿瘤可位于任何器官或组织,包括肺、结肠、肾脏、胰腺、肝脏、骨、皮肤、或肠。脂质体纳米粒子可由可离子化脂质、结构脂质、一种或多种脂质稳定剂、和用于降低纳米粒子的免疫原性的脂质构成。可离子化脂质可选自由化合物81、化合物71、化合物57、化合物84、化合物49,化合物76、化合物78、和化合物102组成的组。本发明还包括用于预防、治疗或改善有需要的受试者的恶性肿瘤的一种或多种症状的方法。所述方法可包括向受试者施予有效量的上述组合物。在一些实施方式中,恶性肿瘤可能与kras突变相关,而上述方法还包括对受试者的肿瘤细胞进行鉴定,所述肿瘤细胞具有以下至少一者:(i)kras基因突变、和(ii)kras蛋白表达水平异常。在某些实施方式中,恶性肿瘤可过量表达gst-π。rnai分子可降低受试者中的gst-π和p21的表达。在一些实施方式中,上述施予可使受试者中的gst-π和p21的表达降低至少5%、且持续至少5天。上述施予还可使受试者的恶性肿瘤的体积缩小至少5%、或至少10%、或至少20%、或至少>30%、或至少>40%、或至少>50%。本申请的方法可减轻恶性肿瘤的一种或多种症状,或者延迟或终止恶性肿瘤的发展。在某些实施方式中,施予可减缓受试者的恶性肿瘤细胞的生长。施予可使受试者的恶性肿瘤细胞的生长减缓至少2%、或至少5%、或至少10%、或至少15%、或至少20%。在一些实施方式中,恶性肿瘤可为肠癌、胰腺癌、肾癌、肺癌、乳腺癌、纤维肉瘤、肺腺癌、粘液腺癌、胰腺导管癌、或结肠直肠癌。本发明的实施方式可提供下述方法,所述方法中,每天施予12次。所述施予可持续1、2、3、4、5、6或7天,或持续1、2、3、4、5、6、8、10或12周。在一些实施方式中,以0.01~2mg/kg的量施予rnai分子,每天至少1次,持续期间为12周以下。在其他实施方式中,对于施予而言,gst-πrnai分子的平均auc(o-last)为1~1000ug*min/ml,平均cmax为0.1~50ug/ml。在某些实施方式中,对于所述施予而言,p21rnai分子的平均auc(o-last)为1~1000ug*min/ml,平均cmax为0.1~50ug/ml。施予方法可以为静脉注射、皮内注射、皮下注射、肌肉内注射、腹腔内注射、口服、局部、输液、或吸入。附图说明图1是表示使用本发明的gst-π和p21的sirna制剂而使癌异种移植肿瘤在体内显著缩小的图。在以脂质体制剂的形式施予至癌异种移植肿瘤的情况下,gst-π和p21的sirna在体内发挥了基因敲低效能。利用的癌异种移植模型被施予了相对较低的量(gst-πsirna为1.15mg/kg,p21sirna为0.74mg/kg)。gst-π和p21的sirna制剂在施予后数天内呈现了显著的肿瘤抑制效果。30天后,gst-π和p21的sirna呈现了明显有利的肿瘤抑制效果,较之对照而言,肿瘤体积缩小2倍。图2是表示使用本发明的gst-π和p21的sirna制剂而使体内癌异种移植肿瘤显著缩小的图。在以脂质体制剂的形式施予至癌异种移植肿瘤的情况下,gst-π和p21的sirna在体内发挥了基因敲低效能。利用的癌异种移植模型被施予了相对较低的量(各sirna为0.75mg/kg)。gst-π和p21的sirna制剂在施予后数天内呈现了显著的肿瘤抑制效果。30天后,gst-π和p21的sirna呈现了明显有利的肿瘤抑制效果,较之对照而言,肿瘤体积缩小1.7倍。图3是表示本发明的gst-π和p21的sirna制剂在体外展示出由癌细胞凋亡导致的癌细胞死亡上升的图。体外癌细胞凋亡的监控通过观察puma的表达上调进行,puma是细胞凋亡的生物标志物,其与细胞活性的下降相关。如图3所示,在gst-π和p21的sirna转染后2~4天,gst-π和p21的sirna制剂使puma的表达水平显著升高。图4是表示本发明的以gst-π作为靶标的sirna导致体内原位肺癌肿瘤显著缩小的图。以脂质体制剂的形式向患有原位a549肺癌肿瘤的无胸腺裸鼠施予2mg/kg的gst-πsirna。对治疗组和介质对照组进行剖检,测定原发瘤的最终重量。在为期6周的研究中,gst-πsirna呈现了显著的肺癌肿瘤抑制效果。如图4所示,43天后,gst-πsirna呈现了明显有利的肿瘤抑制效果,较之对照而言,原发瘤的最终平均重量大幅度减轻2.8倍。图5是表示gst-πsirna的体内肿瘤抑制效果的图。向使用a549细胞的癌异种移植模型以相对较低的量(0.75mg/kg)施予sirna。gst-πsirna在数天内呈现了有利的肿瘤抑制。36天后,gst-πsirna呈现了明显有利的肿瘤抑制,较之对照而言,肿瘤的最终平均体积显著缩小2倍左右。图6是表示本发明的gst-πsirna使由体内凋亡导致的癌细胞死亡显著上升的图。gst-πsirna导致了puma表达上调,puma是细胞凋亡的生物标志物,其与细胞活性的下降相关。图6中,在gst-πsirna转染2~6天后,puma的表达显著升高。图7是表示本发明的gst-πsirna在体内对于a549异种移植肿瘤发挥了敲低效果的图。在施予了以gst-π作为靶标的sirna的无胸腺裸(nu/nu)雌鼠(charlesriver)中观察到gst-πmrna的剂量依赖性敲低。如图7所示,在以4mg/kg注射24小时后,观测到gst-πmrna显著下降约40%。具体实施方式本发明提供递送至恶性肿瘤的治疗组合物中使用的化合物和组合物。在一些方面,本发明涉及用于提供纳米粒子的化合物、组合物和方法,所述纳米粒子用于将活性剂或药物化合物递送并分配至恶性肿瘤细胞、及组织、器官、和患有恶性肿瘤的受试者。本发明提供一系列用于将活性剂递送至恶性肿瘤细胞的可离子化化合物。除了其他用途以外,本发明的可离子化化合物可用于形成递送及分配用于改善恶性肿瘤的活性剂的纳米粒子。本发明的实施方式包括广范围的具有与脂质相似性质的化合物,这些化合物可用于递送使恶性肿瘤细胞摄取的活性剂。本发明的组合物和方法可用于分配基因表达阻遏剂。基因表达阻遏剂的例子包括抑制核酸分子,例如,核酶、反义核酸、和rna干扰分子(rnai分子)。另一方面,本发明提供利用使gst-π核酸分子或多肽、和p21核酸分子或多肽的表达降低的治疗性组合物来治疗受试者的肿瘤的方法,所述方法中,肿瘤与具有kras突变或呈现异常的kras表达水平的细胞有关。本发明的治疗性组合物可含有抑制核酸分子,例如,sirna、shrna、反义rna,以及dna指导的rna(ddrna)、piwi-相互作用rna(pirna)、和重复相关sirna(rasirna)。本文中,kras相关恶性肿瘤或kras相关癌定义为:(a)具有体细胞kras突变的癌细胞或肿瘤细胞、或(b)kras表达水平异常的癌细胞或肿瘤细胞,所述表达水平异常包括但不限于kras编码dna的扩增、或者kras基因较之正常的非癌细胞的水平而言过量表达或低表达。gst-π表示由gstp1基因编码的酶,其催化谷胱甘肽的缀合反应。gst-π存在于包括人在内的各种动物中,其序列信息是已知的,并可提供ncbi数据库登录号(例如,人:np_000843(nm_000852),大鼠:np_036709(nm_012577),小鼠:np_038569(nm_013541)等)。本发明包括用于阻遏编码gst-π的dna的rnai分子、核酶,反义核酸、dna/rna嵌合多核苷酸、用于表达它们的载体、和gst-π的显性失活变体。p21存在于包括人在内的各种动物中,其序列信息也是已知的(例如,人:nm_000389.4、nm_078467.2、nm001291549.1、nm_001220778.1、nm_001220777.1(np_001207707.1、ρ_001278478.1、np_001207706.1、p_510867.1、p_000380.1)等,上述编号代表ncbi数据库登录号,括号内外分别表示氨基酸序列和核苷酸序列)。例如,人cdkn1a基因的核苷酸序列在数据库中登录为m_000389.4。对于p21而言,其序列信息已被登录为上述多个登录号,并且存在多个转录变体。本发明包括用于阻遏编码p21的dna的rnai分子、核酶、反义核酸、dna/rna嵌合多核苷酸、用于表达它们的载体、和p21的显性失活变体。通常,在受试者被诊断为患有与kras突变或kras扩增相关的肿瘤(例如,肺癌、肾癌或胰腺癌)后,可选择阻遏抑制gst-π和p21的治疗方法。此处使用的gst-π阻遏剂的例子包括阻遏gst-π生产及/或活性的药物、促进gst-π分解及/或失活的药物。阻遏gst-π生产的药物的例子包括rnai分子、核酶、反义核酸、gst-π编码dna的dna/rna嵌合多核苷酸、或表达它们的载体。此处使用的p21阻遏剂的例子包括阻遏p21生产及/或活性的药物、促进p21分解及/或失活的药物。阻遏p21生产的药物的例子包括rnai分子、核酶、反义核酸、p21编码dna的dna/rna嵌合多核苷酸、或表达它们的载体。rnai分子本领域技术人员可以理解,已提交的序列可随时间而变更,以引入核酸分子中任何需要的改变。本发明的实施方式可提供使用核酸分子的组合物和方法,其用于gst-π基因表达沉默。本发明的实施方式可提供使用核酸分子的组合物和方法,其用于p21基因表达沉默。本发明的实施方式可提供使用核酸分子的组合物和方法,其用于同时使gst-π和p21基因的表达沉默。能够介导rna干扰的核酸分子的例子包括具有rna干扰活性的分子(rnai分子),其包括rna双链体、例如sirna(小干扰rna)、mirna(微rna)、shrna(短发夹rna)、ddrna(dna指导的rna)、pirna(piwi-相互作用rna)、或rasirna(重复相关sirna)、和它们的修饰体。本文中公开的组合物和方法也可用于治疗受试者的各种恶性肿瘤。为了下调gst-π编码基因的表达、及p21编码基因的表达,可将本发明的核酸分子和方法集中或组合使用。本发明的组合物和方法可包含一种或多种核酸分子,通过将所述核酸分子组合使用,能够调节或调控gst-π和p21蛋白及/或蛋白编码基因的表达、与疾病维持及/或发展相关的蛋白及/或蛋白编码基因的表达、以及例如恶性肿瘤等与gst-π和p21有关的紊乱或病症。参考gst-π和p21的示例性序列,对本发明的组合物和方法进行说明。本领域技术人员可以理解,本发明的各方面和实施方式可对应任何相关的gst-π或p21的基因、序列、或变体(例如,同源基因和转录变体)、以及多态性、例如与任何gst-π或p21基因相关的单核苷酸多态性(snp)。在一些实施方式中,本发明的组合物和方法可提供下调gst-π基因(例如,人gst-π)表达的双链短干扰核酸(sirna)分子。本发明的组合物和方法还可提供下调p21基因(例如,人p21)表达的双链短干扰核酸(sirna)分子。本发明的rnai分子以gst-π或p21、和任何能够提供其他靶序列的同源序列作为靶标,例如使用互补序列或通过并入非典型(canonical)碱基对例如错配和/或摆动(wobble)碱基对(其可提供额外的靶序列)。在鉴定错配的情况下,可以使用非典型碱基对,例如错配和/或摆动碱基来产生靶向多于一个基因序列的核酸分子。例如,uu和cc碱基等非常规碱基对可用于生成能以具有序列同源性的不同的靶序列作为靶标的核酸分子。由此,rnai分子可将同源基因的保守核苷酸序列作为靶标,从而可使用1种rnai分子抑制多个基因的表达。在一些方面,本发明的组合物和方法包含对于gst-πmrna的任何部分具有干扰活性的rnai分子。所述rnai分子可含有与任何编码gst-π的mrna序列互补的序列。本发明还涉及包含对于p21mrna的任何部分具有干扰活性的rnai分子的组合物和方法。所述rnai分子可含有与任何编码p21的mrna序列互补的序列。在一些实施方式中,本申请公开的rnai分子可具有对于gst-πrna的干扰活性,所述rnai分子含有与具有gst-π变体编码序列(例如,本领域已知的与恶性肿瘤相关的gst-π基因突变)的rna互补的序列。在一些实施方式中,本申请公开的rnai分子可具有对于p21rna的干扰活性,所述rnai分子含有与具有p21变体编码序列(例如,本领域已知的与恶性肿瘤相关的p21基因突变)的rna互补的序列。在另一些实施方式中,本发明的rnai分子可含有能够介导gst-π基因表达沉默的核苷酸序列。在另一些实施方式中,本发明的rnai分子可含有能够介导p21基因表达沉默的核苷酸序列。本发明的以gst-πmrna作为靶标的rnai分子的例子示于表1。表1:gst-πrnai分子序列表1备注:大写字母a、g、c、u分别表示ribo-a、ribo-g、ribo-c和ribo-u。小写字母a、u、g、c、t分别表示2’-脱氧-a、2’-脱氧-u、2’-脱氧-g、2’-脱氧-c和脱氧胸苷。本发明的以gst-πmrna作为靶标的rnai分子的例子示于表2。表2:gst-πrnai分子序列表2备注:大写字母a、g、c、u分别表示ribo-a、ribo-g、ribo-c和ribo-u。小写字母a、u、g、c、t分别表示2’-脱氧-a、2’-脱氧-u、2’-脱氧-g、2’-脱氧-c和脱氧胸苷(dt=t=t)。下划线表示被2’-ome取代,例如,u。小写字母f表示2’-脱氧-2’-氟代,例如,fu为2’-脱氧-2’-氟-u。n表示a、c、g、u、u、a、c、g、u、t、或者经过修饰、反转、或化学修饰的核苷酸。本发明的以gst-πmrna作为靶标的rnai分子的例子示于表3。表3:gst-πrnai分子序列表3备注:大写字母a、g、c、u分别表示ribo-a、ribo-g、ribo-c和ribo-u。小写字母a、u、g、c、t分别表示2’-脱氧-a、2’-脱氧-u、2’-脱氧-g、2’-脱氧-c和脱氧胸苷(dt=t=t)。下划线表示被2’-ome取代,例如,u。小写字母f表示2’-脱氧-2’-氟代,例如,fu为2’-脱氧-2’-氟-u。n表示a、c、g、u、u、a、c、g、u、t、或者经过修饰、反转、或化学修饰的核苷酸。本发明的以gst-πmrna作为靶标的rnai分子的例子示于表4。表4:gst-πrnai分子序列表4备注:大写字母a、g、c、u分别表示ribo-a、ribo-g、ribo-c和ribo-u。小写字母a、u、g、c、t分别表示2’-脱氧-a、2’-脱氧-u、2’-脱氧-g、2’-脱氧-c和脱氧胸苷(dt=t=t)。下划线表示被2’-ome取代,例如,u。小写字母f表示2’-脱氧-2’-氟代,例如,fu为2’-脱氧-2’-氟-u。n表示a、c、g、u、u、a、c、g、u、t、或者经过修饰、反转、或化学修饰的核苷酸。本发明的以gst-πmrna作为靶标的rnai分子的例子示于表5。表5:gst-πrnai分子序列表5备注:大写字母a、g、c、u分别表示ribo-a、ribo-g、ribo-c和ribo-u。小写字母a、u、g、c、t分别表示2’-脱氧-a、2’-脱氧-u、2’-脱氧-g、2’-脱氧-c和脱氧胸苷(dt=t=t)。下划线表示被2’-ome取代,例如,u。小写字母f表示2’-脱氧-2’-氟代,例如,fu为2’-脱氧-2’-氟-u。n表示a、c、g、u、u、a、c、g、u、t、或者经过修饰、反转、或化学修饰的核苷酸。本发明的以gst-πmrna作为靶标的rnai分子的例子示于表6。表6:gst-πrnai分子序列表6备注:大写字母a、g、c、u分别表示ribo-a、ribo-g、ribo-c和ribo-u。小写字母a、u、g、c、t分别表示2’-脱氧-a、2’-脱氧-u、2’-脱氧-g、2’-脱氧-c和脱氧胸苷(dt=t=t)。下划线表示被2’-ome取代,例如,u。小写字母f表示2’-脱氧-2’-氟代,例如,fu为2’-脱氧-2’-氟-u。n表示a、c、g、u、u、a、c、g、u、t、或者经过修饰、反转、或化学修饰的核苷酸。本发明的以p21mrna作为靶标的rnai分子的例子示于表7。表7:p21rnai分子序列表7备注:大写字母a、g、c、u分别表示ribo-a、ribo-g、ribo-c和ribo-u。小写字母a、g、c、t分别表示2’-脱氧-a,2’-脱氧-g,2’-脱氧-c和胸苷。mu为2’-甲氧基-u。本发明的以p21mrna作为靶标的rnai分子的例子示于表8。表8:p21rnai分子序列表8备注:大写字母a、g、c、u分别表示ribo-a、ribo-g、ribo-c和ribo-u。小写字母a、u、g、c、t分别表示2’-脱氧-a、2’-脱氧-u、2’-脱氧-g、2’-脱氧-c和脱氧胸苷(dt=t=t)。下划线表示被2’-ome取代,例如,u。n表示a、c、g、u、u、a、c、g、u、t、或者经过修饰、反转、或化学修饰的核苷酸。在一些实施方式中,本发明提供一系列核酸分子,其中,a)所述分子具有多核苷酸有义链和多核苷酸反义链;b)所述分子的每条链长度为15~30个核苷酸;c)反义链中的15~30个核苷酸的连续区域与编码p21的mrna序列互补;以及d)有义链的至少一部分与反义链的至少一部分互补,并且,所述分子具有长度为15~30个核苷酸的双链区。在一些实施方式中,核酸分子可在反义链具有15~30个核苷酸的连续区域,所述区域与编码p21的mrna序列互补,并且位于分子的双链区。在另一些实施方式中,核酸分子可在反义链具有15~30个核苷酸的连续区域,并且,所述区域的序列与编码p21的mrna序列互补。在其他方面,本发明的核酸分子的每条链的长度可以为18~22个核苷酸。核酸分子还可具有长度为19个核苷酸的双链区。在某些实施方式中,核酸分子可具有多核苷酸有义链和多核苷酸反义链,所述有义链和反义链以单链形式连接,并且形成一端以环(loop)连接的双链区。本发明的核酸分子可具有平末端,也可具有一个或多个3’悬挂。本发明的核酸分子可以是具有基因沉默活性的rnai分子,例如,具有基因沉默活性的dsrna、sirna、micro-rna、或具有基因沉默活性的shrna、及dna指导的rna(ddrna)、piwi-相互作用rna(pirna)、重复相关sirna(rasirna)。本发明提供一系列具有p21表达抑制活性的核酸分子。在一些实施方式中,核酸分子的p21敲低的ic50可小于100pm。在另一些实施方式中,核酸分子的p21敲低的ic50可小于50pm。本发明还包括含有一种或多种本发明的核酸分子和药学上允许的担体的组合物。所述担体可以是脂质分子或脂质体。对于本发明的化合物和组合物而言,通过将其施予至需要的受试者,所述化合物和组合物对于p21相关疾病的预防或治疗的方法有用。在其他方面,本发明包括p21表达相关疾病的治疗方法,所述方法中,向有需要的受试者施予含有一种或多种本发明的核酸分子的组合物。所述疾病可以是恶性肿瘤,其可出现于与sp21表达相关的癌症等疾病中。在一些实施方式中,本发明提供一系列核酸分子,其中,a)所述分子具有多核苷酸有义链和多核苷酸反义链;b)所述分子的每条链长度为15~30个核苷酸;c)反义链中的15~30个核苷酸的连续区域与编码gst-π的mrna序列互补;d)有义链的至少一部分与反义链的至少一部分互补,并且,分子具有长度为15~30个核苷酸的双链区。在一些实施方式中,核酸分子可在反义链具有15~30个核苷酸的连续区域,所述区域与编码gst-π的mrna序列互补,并且位于分子的双链区。在另一些实施方式中,核酸分子可在反义链具有15~30个核苷酸的连续区域,并且,所述区域与序列编码gst-π的mrna序列互补。在某些实施方式中,核酸分子的每条链的长度可以为18~22个核苷酸。核酸分子的双链区长度可以为19个核苷酸。在一些替代形式中,核酸分子可具有多核苷酸有义链和多核苷酸反义链,所述有义链和反义链以单链形式连接,并且形成一端以环连接的双链区。在一些实施方式中,本申请公开的核酸分子可具有平末端。在某些实施方式中,核酸分子可具有一个或多个3’悬挂。本发明提供一系列具有基因沉默活性的核酸分(即rnai分子)。本发明的核酸分子可以是dsrna、sirna、micro-rna、或具有基因沉默活性的shrna、及dna指导的rna(ddrna)、piwi-相互作用rna(pirna)、或重复相关sirna(rasirna)。核酸分子可具有gst-π表达抑制活性。本发明的实施方式还提供gst-π敲低的ic50小于100pm的核酸分子。本发明的另一些实施方式提供gst-π敲低的ic50小于50pm的核酸分子。本发明还包括含有一种或多种本发明的核酸分子和药学上允许的担体的组合物。在某些实施方式中,所述担体可以是脂质分子或脂质体。对于本发明的化合物和组合物而言,通过将其施予至需要的受试者,所述化合物或组合物对于gst-π相关疾病的预防或治疗的方法有用。本文中,rnai分子指任何能够导致rna干扰的分子,其包括rna双链体,例如,sirna(小干扰rna)、mirna(微rna)、shrna(短发夹rna)、ddrna(dna指导的rna)、pirna(piwi-相互作用rna)、或rasirna(重复相关sirna)、和它们的修饰体。这些rnai分子可通过购买获得,或根据已知的序列信息进行设计或制备等。反义核酸包含rna、dna、pna、或它们的复合物。本文中,dna/rna嵌合多核苷酸包含由抑制靶基因表达的dna和rna构成的双链多核苷酸。在一个实施方式中,本发明的制剂含有sirna作为治疗剂。sirna分子的长度可为10~50个以上核苷酸。sirna分子的长度可为15~45个核苷酸。sirna分子的长度可为19~40个核苷酸。sirna分子的长度可为19~23个核苷酸。本发明的sirna分子能够介导针对靶mrna的rnai。sirna的设计和生产可利用市售的设计工具和试剂盒进行,例如,可使用ambion,inc.(德克萨斯州奥斯汀市)、麻省理工学院怀特黑德生物医学研究所(马萨诸塞州剑桥市)的制品。恶性肿瘤的治疗方法本发明的实施方式可提供能够用于下调或抑制gst-π及/或gst-π蛋白的表达的rnai分子,以及能够用于下调或抑制p21及/或p21蛋白的表达的rnai分子。在一些实施方式中,本发明的rnai分子可用于下调或抑制gst-π及/或gst-π蛋白的表达,所述gst-π及/或gst-π蛋白源于可能与例如恶性肿瘤等疾病或病症相关的gst-π单倍型多态性。本发明的实施方式还包含用于下调或抑制p21及/或p21蛋白表达的rnai分子,所述p21及/或p21蛋白源于可能与例如恶性肿瘤等疾病或症状相关的p21单倍型多态性。对于gst-π的蛋白或mrna的水平、及p21的蛋白或mrna的水平的监控可用于表征基因沉默,以及测定本发明的化合物和组合物的效果。本申请公开的rnai分子可单独使用,也可与其他sirna组合使用来调节一个或多个基因的表达。本申请公开的rnai分子可单独使用,也可与其他已知的药物组合或缀合使用,由此预防或治疗疾病、或改善恶性肿瘤等与gst-π和p21相关的病症或紊乱的症状。本发明的rnai分子可用于以序列特异性方式调节或抑制gst-π表达。另外,本发明的rnai分子可用于以序列特异性方式调节或抑制p21表达。本申请公开的rnai分子可包含引导链(guidestrand),其是一系列连续的核苷酸,至少部分与gst-πmrna或p21mrna互补。在特定的方面,可使用本发明的rnai分子通过rna干扰对恶性肿瘤进行治疗。对于恶性肿瘤的治疗而言,可利用合适的以细胞为基础的模型、动物模型(体外或体内)进行表征。对于恶性肿瘤的治疗而言,可通过测定受影响组织的细胞中的gst-πmrna水平或gst-π蛋白水平进行表征。对于恶性肿瘤的治疗而言,可通过测定受影响组织的细胞中的p21mrna水平或p21蛋白水平进行表征。对于恶性肿瘤的治疗而言,可通过对受影响的器官或组织进行非侵入性的医学扫描进行表征。本发明的实施方式可包括对有需要的受试者的gst-π及/或p21相关疾病或病症的症状进行预防、治疗、或改善的方法。在一些实施方式中,对受试者的恶性肿瘤的症状进行预防、治疗、或改善的方法可包括向受试者施予本发明的rnai分子,由此调节受试者或生物体中的gst-π基因及/或p21基因的表达。在一些实施方式中,本发明包括下调细胞或生物体中的gst-π基因表达的方法,所述方法中,使细胞或生物体与本发明的rnai分子接触。本发明还包括下调细胞或生物体中的p21基因表达的方法,所述方法中,使细胞或生物体与本发明的rnai分子接触。抑制gst-π和p21的核酸分子可以是使基因表达降低的寡核苷酸,其可以采用单链或双链核酸分子结构。在一个方案中,抑制核酸分子为用于rna干扰(rnai)介导基因表达敲低的双链rna。在一个实施方式中,具有rna干扰活性的rna双链体(dsrna)分子含有本发明的寡核苷酸中的8~25(例如、8、10、12、15、16、17、18、19、20、21、22、23、24、25)个连续的核苷酸。dsrna可以是2条形成双链结构的互补rna链,也可以是1条自体形成双链结构的rna链(短发夹(sh)rna)。在一些实施方式中,具有rna干扰活性的dsrna具有约21或22个碱基对,也可以更长或更短,最长为29个核苷酸。双链rna可以利用常规技术制备,例如,化学合成或体外转录。也可使用试剂盒,例如,由ambion(德克萨斯州奥斯汀市)和epicentre(威斯康星州麦迪逊市)提供的试剂盒。在哺乳动物细胞中表达dsrna的方法记载于以下文献:brummelkampetal.science296:550-553,2002;paddisonetal.genes&devel.16:948-958,2002;pauletal.naturebiotechnol.20:505-508,2002;suietal.,proc.natl.acad.sci.usa99:5515-5520,2002;yuetal.proc.natl.acad.sci.usa99:6047-6052,2002;miyagishietal.,naturebiotechnol.20:497-500,2002;及leeetal.,naturebiotechnol.20:500-5052002,其内容通过参考并入本说明书中。与gst-π基因“对应”的抑制核酸分子含有至少1段双链基因,由此,双链抑制核酸分子的每条链能够与gst-π靶基因的互补链结合。抑制核酸分子不需要与gst-π参考序列完全一致。与p21基因“对应”的抑制核酸分子含有至少1段双链基因,由此,双链抑制核酸分子的每条链能够与p21靶基因的互补链结合。抑制核酸分子不需要与p21参考序列完全一致。在一个实施方式中,sirna与靶核酸具有至少约85%、90%、95%、96%、97%、98%、99%的序列同一性。例如,19个碱基对的、含有1~2处碱基对错配的双链可用于本发明的方法。在其他的实施方式中,抑制核酸分子的核苷酸序列显示了1、2、3、4、5处或更多的错配。本发明提供的抑制核酸分子不限于sirna,而是包含能降低gst-π或p21的核酸分子或多肽的表达的任何核酸分子。本发明提供的dna序列可用于例如发现和开发能降低编码蛋白表达的治疗性反义核酸分子。本发明还提供催化性rna分子或核酶。这样的催化性rna分子可用于抑制靶核酸分子的体内表达。通过在反义rna中包含核酶序列,能够赋予分子rna切割活性,从而提高构建体的活性。靶rna特异性核酶的设计和利用记载于haseloffetal.,nature334:585-591.1988和us2003/0003469al,其内容通过参考并入本说明书中。在本发明的各实施方式中,催化性核酸分子形成锤头状或发夹状基序(motif)。上述锤头基序的例子记载于rossietal.,aidsresearchandhumanretroviruses,8:183,1992。上述发夹基序的例子记载于hampeletal.,biochemistry,28:4929,1989,andhampeletal.,nucleicacidsresearch,18:299,1990。本领域技术人员熟知,对于酶性核酸分子而言,特异性底物结合位点是必须的,所述位点与一个或多个靶基因rna区域互补,并且,所述位点内部或附近的核苷酸序列能够赋予分子rna切割活性。对靶标的阻遏可通过比较相应蛋白在被阻遏的细胞和未使用阻遏剂的细胞中的表达或活性来测定。蛋白表达可利用任何已知的技术进行评价,例如,利用抗体的免疫沉淀法、eia、elisa、ira、irma、western印迹法、免疫组织化学方法、免疫细胞化学方法、流式细胞术、各种利用与编码蛋白的核酸或其特有片段、或者所述核酸的转录产物(例如,mrna)或剪接产物的特异性杂交的杂交法、northern印迹法、southern印迹法、以及各种pcr方法。蛋白活性可通过分析所述蛋白的已知活性进行评价,所述活性包括与例如raf-1(尤其是磷酸化raf-1)或egfr(尤其是磷酸化egfr)等结合,所述评价可利用任何已知方法,例如,免疫沉淀法、western印迹法,amass分析、下拉法(pull-down)、或表面等离子体共振(spr)法。kras突变体的例子包括但不限于导致持续激活kras的突变,例如,抑制内源性gtpase的突变或提高鸟嘌呤核苷酸交换率的突变。这样的突变的具体例子包括但不限于:例如,人kras第12、13及/或61位氨基酸突变(抑制内源性gtpase)、和人kras第116及/或119位氨基酸突变(提高鸟嘌呤核苷酸交换率)(bos,cancerres.1989;49(17):4682-9,levietal.,cancerres.1991;51(13):3497-502)。在本发明的一些实施方式中,kras突变体可以是在人kras第12、13、61、116、119位氨基酸中的至少一处发生突变的kras。在本发明的一个实施方式中,kras突变体在人kras第12位氨基酸发生突变。在一些实施方式中,kras突变体可能会诱导gst-π的过量表达。具有kras突变的细胞可能会呈现gst-π的过量表达。可利用任何已知的技术对kras突变进行检测,例如,利用对于已知突变序列具有特异性的核酸探针进行的选择性杂交、酶促错配切割法、测序(bos,cancerres.1989;49(17):4682-9)、pcr-rflp法(miyanishietal.,gastroenterology。2001;121(4):865-74).)。可利用任何已知的技术对靶表达进行检测。为了评价该靶标是否过量表达,例如,可将具有kras突变的细胞中的靶表达程度与含有正常kras的同型细胞中的靶表达程度进行比较。该情况下,如果具有kras突变的细胞中的靶表达程度高于含有正常kras的同型细胞中的靶表达程度,则评价为过量表达。在一个方面,本发明的特征在于,含有编码上述任一种抑制核酸分子的载体。在具体的实施方式中,载体为逆转录病毒载体、腺病毒载体、腺相关病毒载体、或慢病毒载体。在另一个实施方式中,载体含有适于哺乳动物细胞中表达的启动子。本发明的组合物中,诱导rna干扰的活性成分的配合量可以为使施予利大于弊的量。所述配合量可通过使用培养细胞的体外试验、或者模型动物或例如小鼠、大鼠、狗、猪等哺乳动物的试验来确定,并且,可使用本领域技术人员熟知的试验方法。本发明方法可应用于包括人在内的任何动物。活性成分的配合量可根据制剂或组合物的施予方式而变化。例如,一次施予中使用多个组合物单位(unit)的情况下,通过将一次施予所需的活性成分量除以所述单位的数量,则可确定每1个单位中的活性成分配合量。本发明还涉及生产用于阻遏gst-π和p21的制剂或组合物的步骤,以及为使恶性肿瘤减轻或缩小而使用阻遏gst-π和p21的组合物的步骤。rna干扰rna干扰(rnai)是指由短干扰rna(sirna)介导的动物中的序列特异性转录后基因沉默。例如,zamoreetal.,cell,2000,vol.101,pp.25-33;fireetal.,nature,1998,vol.391,pp.806811;sharp,genes&development,1999,vol.13,pp.139-141。细胞中的rnai反应可以由双链rna(dsrna)引发,其机理尚未彻底研明。在细胞中,作为rnaseiii酶的dicer酶可作用于特定的dsrna。例如,参见zamoreetal.,cell,2000,vol.101,pp.25-33;hammondetal.,nature,2000,vol.404,pp.293-296。dicer可将dsrna加工成更短的dsrna片段,即sirna。一般而言,sirna的长度可以为21~约23个核苷酸,并且包含长度为约19个核苷酸的碱基对双链区。rnai涉及被称为rna诱导沉默复合物(risc)的核酸内切酶复合物。sirna具有的反义链或引导链进入risc复合物,并介导单链靶rna的切割,所述单链靶rna含有与sirna双链体的反义链互补的序列。sirna的另一条链为随从链(passengerstrand)。靶rna的切割在与sirna双链体的反义链互补的区域中部发生。例如,参见elbashiretal.,genes&development,2001,vol.15,pp.188-200。本文中,用语“有义链”表示sirna分子的下述核苷酸序列,所述序列与sirna分子的反义链的至少一部分完全互补或部分互补。sirna分子的有义链可含有与靶核酸序列具有同源性的核酸序列。本文中,用语“反义链”表示与靶核酸序列的至少一部分完全互补或部分互补的sirna分子的核苷酸序列。sirna分子的反义链可含有与该sirna分子的有义链的至少一部分互补的核酸序列。rnai分子可通过以序列特异性方式介导rna干扰来下调或敲低基因表达。例如,参见zamoreetal.,cell,2000,vol.101,pp.25-33;elbashiretal.,nature,2001,vol.411,pp.494-498;kreutzeretal.,wo2000/044895;zernicka-goetzetal.,wo2001/36646;fireetal.,wo1999/032619;plaetincketal.,wo2000/01846;melloetal.,wo2001/029058。本文中,关于基因表达的用语“抑制”、“下调”或“降低”是指,观察到基因表达、或编码一种或多种蛋白的mrna分子的水平、或一种或多种编码蛋白的活性低于不存在本发明的rnai分子或sirna的情况。例如,可观察到较之不存在本发明的rnai分子或sirna的情况而言,表达水平、mrna水平、或编码蛋白的活性水平降低至少1%、或至少10%、或至少20%、或至少50%、或至少90%以上。rnai分子还可用于敲低病毒基因表达,从而能够影响病毒复制。rnai分子可以由独立的多核苷酸链制得:有义链或随从链、和反义链或引导链。引导链与随从链至少部分互补。引导链和随从链可形成具有约15~49个碱基对的双链区。在一些实施方式中,sirna双链区可具有17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、或49个碱基对。在某些实施方式中,rnai分子可在risc复合物中具有活性,其具有对于ric有活性的双链体区域长度。在另一些实施方式中,rnai分子可以作为dice底物发挥作用,并被转换为能够在risc复合物中发挥作用的rnai分子。在一些方面,rnai分子可在长链分子相反的两端具有互补的引导序列部分和随从序列部分,由此,分子能够介由互补的序列部分而形成双链区,在双链区的一端介由核苷酸接头或非核苷酸接头将链彼此连接。例如,发夹结构或茎环结构。接头与链的相互作用可以是共价键或非共价相互作用。本申请公开的rnai分子可包含将核酸的有义区域与反义区域接合的核苷酸接头、非核苷酸接头、或核苷酸/非核苷酸混合接头。核苷酸接头的长度可以为≥2个核苷酸,例如约3、4、5、6、7、8、9、或10个核苷酸。核苷酸接头可以是核酸适体。本文中,用语“适体”或“核酸适体”表示与靶分子特异性结合的核酸分子,所述核酸分子的序列中包含天然能够被靶分子识别的序列。或者,适体也可以是天然不会与核酸结合的靶分子结合的核酸分子。例如,适体可用于与蛋白质的配体结合区域结合,由此阻碍天然存在的配体与蛋白的相互作用。例如,参见goldetal.,annurevbiochem,1995,vol.64,pp.763-797;brodyetal.,j.biotechnol.,2000,vol.74,pp.5-13;hermannetal.,science,2000,vol.287,pp.820-825。非核苷酸接头的例子包括脱碱基核苷酸、聚醚、聚胺、聚酰胺、肽、碳水化合物、脂质、聚烃、或其他聚合物,例如,具有例如2~100个乙二醇单元的聚乙二醇。一些例子记载于seelaetal.,nucleicacidsresearch,1987,vol.15,pp.3113-3129;cloadetal.,j.am.chem.soc.,1991,vol.113,pp.6324-6326;jaeschkeetal.,tetrahedronlett.,1993,vol.34,pp.301;arnoldetal.,wo1989/002439;usmanetal.,wo1995/006731;dudyczetal.,wo1995/011910;和ferentzetal.,j.am.chem.soc.,1991,vol.113,pp.4000-4002。rnai分子可在双链区具有一个或多个悬挂。悬挂是碱基未配对的单链区,长度可为1~8个核苷酸或更长。悬挂可以是3’悬挂,其中,链的3’端具有长度为1~8个核苷酸的单链区。悬挂也可以是5’悬挂,其中,链的5’端具有长度为1~8个核苷酸的单链区。rnai分子的悬挂的长度可以相同,也可以不同。rnai分子可具有一个或多个平末端,即,双链区的末端没有悬挂,两条链的碱基配对直到双链区的末端。本申请公开的rnai分子可具有一个或多个平末端,也可具有一个或多个悬挂,或者具有平末端和悬挂的组合。rnai分子链的5’端可以是平末端,或者也可以是悬挂。rnai分子链的3’端可以是平末端,或者也可以是悬挂。rnai分子链的3’端为悬挂时,5’端可以是平末端。rnai分子链的5’端为悬挂时,3’端可以是平末端。在一些实施方式中,rnai分子的两端可均为平末端。在另一些实施方式中,rnai分子的两端可均具有悬挂。5’端和3’端的悬挂的长度可以不同。在某些实施方式中,rnai分子可具有平末端,即,在反义链的5’端和有义链的3’端不具有任何悬挂的核苷酸。在另一些实施方式中,rnai分子可具有平末端,即,在反义链的3’端和有义链的5’端不具有任何悬挂的核苷酸。rnai分子可在双链区具有碱基错配。rnai分子的悬挂中的任意的核苷酸可以是脱氧核糖核苷酸,也可以是核糖核苷酸。rnai分子可在5’端具有一个或多个脱氧核糖核苷酸,另一条链的3’端可不具有悬挂,或不具有脱氧核糖核苷酸悬挂。rnai分子可在3’端具有一个或多个脱氧核糖核苷酸,另一条链的5’端可不具有悬挂,或不具有脱氧核糖核苷酸悬挂。在一些实施方式中,rnai分子的一个或多个、或者全部悬挂的核苷酸可以为2’-脱氧核糖核苷酸。dicer底物rnai分子在一些方面,rnai分子可具有适合作为dizcer底物的长度,并且能够被加工成具有risc活性的rnai分子。例如,参见rossietal.,us2005/0244858。作为dicer底物,双链rna(dsrna)具有对于被dicer加工成活性rnai分子而言充分的长度,并具有以下一种或多种性质:(i)作为dicer底物的dsrna可以是不对称的,例如,可在反义链具有3’悬挂;及(ii)dicer底物dsrna可在有义链的3’末端具有修饰,由此引导dicer结合和加工的方向和dsrna加工成活性rnai分子。rnai分子的使用方法本发明的核酸分子和rnai分子可直接用于递送至细胞或组织,也可与担体或稀释剂结合。对于本发明的核酸分子和rnai分子而言,可使用担体或稀释剂、或任何其他发挥辅助、促进或加快进入细胞的作用的递送介质(vehicle)(例如,病毒序列、病毒物质、或者脂质或脂质体制剂),由此将分子直接用于递送或施予至细胞、组织、器官、或受试者。本发明的核酸分子和rnai分子可以与阳离子性脂质形成复合物、被脂质体包封、或以其它方式递送至靶细胞或靶组织。核酸或核酸复合物可通过直接经皮施用、透皮施用、或注射而局部施予至体外或体内的有关组织。递送体系可包括:例如,水性和非水性的凝胶、乳霜(cream)、乳剂(emulsion)、微乳剂、脂质体、软膏、水性和非水性的溶液、乳液(lotion)、气溶胶(aerosols)、烃类基剂和粉末,并且可含有例如助溶剂、渗透促进剂等赋形剂。可向药学上允许的稀释剂、担体或赋形剂中添加本发明的抑制核酸分子或组合物,以单位剂型进行施予。可采用常规制药操作提供合适的制剂或组合物,由此将化合物施予至患有由细胞过度增殖导致的疾病的患者。施予可在患者显出症状之前开始。施予可采用任何合适的途径,例如,可为胃肠外、静脉、皮下、瘤内、肌肉内、颅内、眼眶内、眼部、心室内、肝内、关节囊内、鞘内、脑池内、腹腔内、鼻内适于、喷雾剂、栓剂、或口服。例如,治疗制剂的剂型可以为液状溶液或悬浮液;对于口服而言,制剂的剂型可以为片剂或胶囊剂;对于鼻内制剂而言,剂型可以为粉剂、滴鼻剂、或喷雾剂。本申请公开的组合物和方法可包含表达载体,所述担体以能够使核酸分子表达的方式而含有编码至少1种本发明的rnai分子的核酸序列。本发明的核酸分子和rnai分子可利用插入dna或rna载体的转录单元表达。重组载体可以是dna质粒或病毒载体。病毒载体可用于使核酸分子短暂表达。例如,载体可含有编码rnai分子双链体的2条链、或自身互补从而能形成rnai分子的单个核酸分子的序列。表达载体可含有编码2个或更多核酸分子的核酸序列。核酸分子可利用真核生物启动子在细胞中表达。本领域技术人员熟知,核酸可利用合适的dna/rna载体而在真核细胞中表达。在一些方面,病毒构建体可用于将表达构建体导入细胞,由此转录该表达构建体编码的、具有rna干扰活性的dsrna构建体。脂质制剂可通过静脉注射、肌肉内注射、腹腔内注射、口服、吸入或本领域已知的其他方法施予至动物。可使用已知能够用于施予寡核苷酸的药学上允许的制剂。在上述方法的一个实施方式中,抑制核酸分子的施予量为约5~500mg/m2/天,例如,可为5、25、50、100、125、150、175、200、225、250、275、或300mg/m2/天。在一些实施方式中,将抑制本发明的核酸分子以1~100mg/kg的用量进行全身施予,例如,为1、5、10、20、25、50、75、或100mg/kg。在另一些实施方式中,用量可为25~500mg/m2/天的范围。制剂制造领域的已知方法可参见例如"remington:thescienceandpracticeofpharmacy"ed.a.r.gennaro,lippincourtwilliams&wilkins,philadelphia,pa.,2000。对于用于胃肠外的施予制剂而言,例如,可含有赋形剂、无菌水、或生理盐水、聚乙二醇等聚亚烷基二醇、植物油、或氢化萘。为了控制化合物的释放,可使用生物相容性、生物降解性的丙交酯聚合物、丙交酯/乙交酯共聚物、或聚氧乙烯-聚氧丙烯共聚物。其他潜在有用的抑制核酸分子的胃肠外递送体系包括乙烯-乙酸乙烯酯共聚物粒子、渗透泵、植入式输液装置、和脂质体。吸入用制剂可包含例如乳糖等赋形剂,或为含有例如聚氧乙烯-9-月桂醚、甘氨胆酸盐和脱氧胆酸盐的水性溶液,或为用于以滴鼻液形式施予的油性溶液、或凝胶。可通过向患者施予治疗有效量(例如,预防、消除或减轻病症的量)的制剂来治疗肿瘤疾病或病症。本发明的寡核苷酸的优选用量取决于下述变量:紊乱的类型和程度、个别患者的总体健康状态、化合物赋形剂的配方、和施予途径。所有上述用于减轻恶性肿瘤的方法可以是体外方法,也可以是体内方法。用量可通过本领域已知的使用培养细胞的体外试验来确定。有效量可以是使kras相关肿瘤的大小缩小至少10%、至少20%、或至少30%、或至少40%、或至少50%、或至少60%、或至少70%、或至少80%、或至少90%、最多为100%的量。本发明的药物组合物能够有效地治疗kras相关疾病。疾病的例子包括由异常细胞增殖导致的疾病、由kras突变导致的疾病、和由gst-π过量表达导致的疾病。由异常细胞增殖导致的疾病的例子包括恶性肿瘤、增生、瘢痕瘤、库欣综合征、原发性醛固酮增多症、黏膜红斑病、真性红细胞增多症、粘膜白斑病、增生性瘢痕、扁平癣、和着色斑病。由kras突变导致的疾病的例子包括恶性肿瘤(也被称为癌症或恶变肿瘤)。由gst-π过量表达导致的疾病的例子包括恶性肿瘤。癌症的例子包括:纤维肉瘤、恶性纤维性组织细胞瘤、横纹肌肉瘤、平滑肌肉瘤、血管肉瘤、卡波西肉瘤、淋巴管肉瘤、滑膜肉瘤、软骨肉瘤、和骨肉瘤等肉瘤;脑癌、头颈癌、乳腺癌、肺癌、食道癌、胃癌、十二指肠癌、阑尾癌、结肠癌、直肠癌、肝癌、肾癌、胰腺癌、胆囊癌、胆管癌、肾细胞癌、输尿管癌、膀胱癌、前列腺癌、睾丸癌、子宫癌、卵巢癌、皮肤癌等上皮性恶性肿瘤;白血病、和恶性淋巴瘤。癌症包括上皮性恶性肿瘤和非上皮性恶性肿瘤。癌可存在于身体的任何部位。本发明的一个实施方式中,癌包括具有上述kras突变的癌细胞。在另一个实施方式中,癌包括呈现不依赖激素或生长因子的增殖的癌细胞。在另一些实施方式中,癌包括呈现gst-π过量表达的癌细胞。用于阻遏gst-π的其他活性剂或药物用于抑制gst-pi活性的其他活性剂的例子包括但不限于:与gst-pi结合的物质,例如,谷胱甘肽、谷胱甘肽类似物(例如,记载于wo95/08563、wo96/40205、w099/54346的物质)、酮洛芬、吲哚美辛(例如,参见halletal.,cancerres.1989;49(22):6265-8)、依他尼酸、吡前列素(例如,参见tewetal.,cancerres.1988;48(13):3622-5)、抗gst-pi抗体、和gst-π显性失活突变体。这些活性剂可通过购买获得,或可基于现有已知技术生产而得到。用于向恶性肿瘤递送活性剂的三元制剂本文中,对于制剂的成分而言,例如,“脂质”可以是单一的化合物,也可以是一种或多种合适的脂质化合物的组合。例如,“脂质稳定剂”可表示单一的稳定剂,或一种或多种合适的脂质稳定剂的组合。本领域技术人员可以容易地理解,无需进行大量试验即可应用本文中记载的特定的化合物组合,以及,各种化合物的组合也属于本文中说明的制剂成分。本发明可提供用于在细胞、组织、器官、或生物体、和受试者中使活性剂分配的组合物,所述组合物含有本发明的一种或多种可离子化脂质分子。本发明的组合物可含有一种或多种可离子化脂质分子、结构脂质、和一种或多种用于降低纳米粒子的免疫原性的脂质。本发明的可离子化脂质分子可以为本发明的组合物的任意mol%。本发明的组合物中,可离子化脂质分子可以为组合物中脂质成分的50mol%~80mol%。在某些实施方式中,组合物中的可离子化脂质分子可以为组合物中脂质成分的55mol%~65mol%。在另一些实施方式中,组合物中的可离子化脂质分子可以为组合物中脂质成分的约60mol%。本发明的组合物中的结构脂质可以为组合物中脂质成分的20mol%~50mol%。在某些实施方式中,组合物中的结构脂质可以为组合物中脂质成分的35mol%~45mol%。一种或多种用于降低纳米粒子的免疫原性的脂质的总量可以为组合物中脂质成分的1mol%~8mol%。在某些实施方式中,一种或多种用于降低纳米粒子的免疫原性的脂质的总量可以为组合物中脂质成分的1mol%~5mol%。在其它方面,本发明的组合物还可含有阳离子性脂质,所述阳离子性脂质可以为组合物中脂质成分的5mol%~25mol%。在某些实施方式中,本发明的组合物还可含有阳离子性脂质,所述阳离子性脂质可以为组合物中脂质成分的5mol%~15mol%。在这些方面,本发明的组合物中的阳离子性脂质与可离子化脂质分子的浓度的摩尔比可以为5:80~25:50。本发明的组合物中,全部脂质成分可包括一种或多种可离子化脂质分子成分、一种或多种结构脂质、和一种或多种用于降低纳米粒子的免疫原性的脂质。用于向恶性肿瘤递送活性剂的四元制剂本发明可提供用于在细胞、组织、器官、或生物体、和受试者中使活性剂分配的组合物,所述组合物含有本发明的一种或多种可离子化脂质分子。本发明的组合物可含有一种或多种可离子化脂质分子、结构脂质、和一种或多种用于降低纳米粒子的免疫原性的脂质。本发明的可离子化脂质分子可以为本发明的组合物的任意mol%。本发明的组合物中,可离子化脂质分子可以为组合物中脂质成分的15mol%~40mol%。在某些实施方式中,组合物中的可离子化脂质分子可以为组合物中脂质成分的20mol%~35mol%。在另一些实施方式中,组合物中的可离子化脂质分子可以为组合物中脂质成分的25mol%~30mol%。本发明的组合物中的结构脂质可以为组合物中脂质成分的25mol%~40mol%。在某些实施方式中,组合物中的结构脂质可以为组合物中脂质成分的30mol%~35mol%。本发明的组合物中,脂质稳定剂的总量可以为组合物中脂质成分的25mol%~40%mol%。在某些实施方式中,脂质稳定剂的总量可以为组合物中脂质成分的30mol%~40mol%。在一些实施方式中,本发明组合物的可含有2种或多种脂质稳定剂,其中,各脂质稳定剂可各自为组合物中脂质成分的5mol%~35mol%。在某些实施方式中,本发明组合物的可含有2种或多种脂质稳定剂,其中,各脂质稳定剂可各自为组合物中脂质成分的10mol%~30mol%。在某些实施方式中,一种或多种脂质稳定剂的总量可以为组合物中脂质的25mol%~40mol%,其中,各脂质稳定剂可各自为5mol%~35%mol%。在某些实施方式中,一种或多种脂质稳定剂的总量可以为组合物中脂质的30mol%~40mol%,其中,各脂质稳定剂可各自为10mol%~30%mol%。一种或多种用于降低纳米粒子的免疫原性的脂质的总量可以为组合物中脂质成分的1mol%~8mol%。在某些实施方式中,一种或多种用于降低纳米粒子的免疫原性的脂质的总量可以为组合物中脂质成分的1mol%~5mol%。在其它方面,本发明的组合物还可含有阳离子性脂质,所述阳离子性脂质可以为组合物中脂质成分的5mol%~25mol%。在某些实施方式中,本发明的组合物还可含有阳离子性脂质,所述阳离子性脂质可以为组合物中脂质成分的5mol%~15mol%。i在这些方面,本发明的组合物中的阳离子性脂质与可离子化脂质分子的浓度的摩尔比可以为5:35~25:15。本发明的组合物中,全部脂质成分可包括一种或多种可离子化脂质分子成分、一种或多种结构脂质、和一种或多种用于降低纳米粒子的免疫原性的脂质。脂质组合物的例子在一些实施方式中,三组脂质类成分,例如,一种或多种可离子化分子、结构脂质、和一种或多种用于降低纳米粒子的免疫原性的脂质可以为组合物中脂质成分的100%。在某些实施方式中,可包含阳离子性脂质。本发明的组合物的例子示于表9。表9:脂质成分的组成(各成分相对于总量的mol%)可离子化阳离子性结构免疫原性降低.600328600355550441650323600364650323700255740206780202501035555152555520205在某些实施方式中,四组脂质类成分,例如,一种或多种可离子化分子、结构脂质、一种或多种脂质稳定剂、和一种或多种用于降低纳米粒子的免疫原性的脂质可以为组合物中脂质成分的100%。本发明的组合物的例子示于表10。表10:脂质成分的组成(各成分相对于总量的mol%)可离子化阳离子性结构稳定剂免疫原性降低.17035408200354052503539125035355250304052504030530025405350253554003025525530355251030305251525305可离子化脂质类分子可离子化脂质的例子包括具有式i表示的结构的化合物。式中,r1和r2为:r1=ch2(ch2)noc(=o)r4r2=ch2(ch2)moc(=o)r5,其中,n和m为1~2;r4和r5在各情况下独立地为c(12-20)烷基、或具有0~2个双键的c(12-20)链烯基;式中,r3选自:其中,r6选自氢原子、烷基、羟基烷基、烷氧基、烷氧烷氧基、氨基烷基;r7选自氢原子、烷基,羟基烷基;q为氧原子或nr7;p为1~4。可离子化脂质的例子包括以下化合物:双(9z,9’z,12z,12’z)-(十八碳-9,12-二烯酸)((2-((3,s’,4r)-3,4-二羟基吡咯烷-l-基)乙酰基)氮烷二基)双(乙烷-2,l-二基)酯。可离子化脂质的例子包括以下化合物:双十四烷酸((2-(3-(羟甲基)杂氮环丁烷-l-基)乙酰基)氮烷二基)双(乙烷-2,l-二基)酯。可离子化脂质的例子包括以下化合物:双(9z,9’z,12z,12’z)-(十八碳-9,12-二烯酸)((2-(4-(2-羟乙基)哌嗪-l-基)乙酰基)氮烷二基)双(乙烷-2,l-二基)酯。可离子化脂质的例子包括以下化合物:双(9z,9’z,12z,12’z)-(十八碳-9,12-二烯酸)((2-(4-(2-羟乙基)哌嗪-l-基)乙酰基)氮烷二基)双(乙烷-2,l-二基)酯。可离子化脂质的例子包括具有式iv表示的结构的化合物。式中,r1和r2为:r1=c(=o)or4r2=c(=o)or5,其中,r4和r5在各情况下独立地为c(12-20)烷基,或具有1~2个双键的c(12-20)链烯基;其中,r3选自:其中,r6选自氢原子、烷基、羟基烷基、烷氧基、烷氧烷氧基、氨基烷基;r7选自氢原子、烷基、羟基烷基;q为氧原子或nr7;p为1~4。可离子化脂质的例子包括以下化合物:2-((l-(((9z,12z)-十七碳-9,12-二烯-1-基)氧基)-5-(((9z,12z)-十八碳-9,12-二烯-1-基)氧基)-1,5-二氧代戊烷-3-基)氨基)-n,n,n-三甲基-2-氧代1-乙铵。可离子化脂质的例子包括具有式vi表示的结构的化合物。式中,r1为:r1=oc(=o)r4其中,r2和r4在各情况下独立地为c(12-20)烷基、或具有1~2个双键的c(12-20)链烯基;其中,r3选自氨基烷基、四级氨基烷基(quternaryaminoalkyl)。可离子化脂质的例子包括以下化合物:(9z,12z)-十八碳-9,12-二烯酸2-((9z,12z)-n-(3-(二甲基氨基)丙基)十八碳-9,12-二烯酰氨基)乙酯。可离子化脂质的例子包括以下化合物:n,n,n-三甲基-3-((9z,12z)-n-(2-(((9z,12z)-十八碳-9,12-二烯酰基)氧基)乙基)十八碳-9,12-二烯酰氨基)-l-丙铵。可离子化脂质的例子包括具有式ix表示的结构的化合物式中,r1和r2为:r1=c(=o)or4r2=nhc(=o)r5其中,r4和r5在各情况下独立地为c(12-20)烷基、或具有1~2个双键的c(12-20)链烯基;其中,r为1~4;其中,r3选自:其中,r6选自氢原子、烷基、羟基烷基、烷氧基、烷氧烷氧基、氨基烷基;r7选自氢原子、烷基、羟基烷基;q为氧原子或nr7。可离子化脂质的例子包括以下化合物:n,n,n-三甲基-2-(((s)-3-(((9z,12z)-十八碳-9,12-二烯-1-基)氧基)-2-((9z,127)-十八碳-9,12-二烯酰氨基)-3-氧代丙基)氨基)-2-氧代-l-乙铵。可离子化脂质分子的例子包括具有式iii表示的结构的化合物。式中,r1和r2为:r1=ch2(ch2)noc(=o)r4r2=ch2(ch2)moc(=o)r5,其中,n和m为1~2;r4和r5在各情况下独立地为c(12-20)烷基、或c(12-20)链烯基;其中,r3选自烷基、羟基烷基、烷氧烷氧基、和羧基烷基;其中,r6选自nr72、n+hr72和n+r73;其中,r7选自氢原子、烷基、羟基烷基。可离子化脂质分子的例子包括具有式iv-b表示的结构的化合物。式中,r1和r2为:r1=c(=o)or4r2=c(=o)or5,其中,r4和r5在各情况下独立地为c(12-20)烷基、或c(12-20)链烯基;其中,z为硫原子或氧原子;其中,r3选自:其中,r6各自独立地选自氢原子、烷基、羟基烷基、烷氧基、烷氧烷氧基、氨基烷基;r7选自氢原子、烷基、羟基烷基;p为1~4。可离子化脂质分子的例子包括具有式v表示的结构的化合物。式中,r1和r2为:r1=nhc(=o)r4r2=c(=o)or5,其中,r4和r5在各情况下独立地为c(12-20)烷基、或c(12-20)链烯基;其中,p为1~4;其中,r3选自:其中,r6各自独立地选自氢原子、烷基、羟基烷基、烷氧基、烷氧烷氧基、氨基烷基;r7选自氢原子、烷基、羟基烷基;q为氧原子或nr7。可离子化脂质分子的例子包括具有式vii表示的结构的化合物。式中,r1和r2为:r1=ch2(ch2)noc(=o)r4r2=ch2(ch2)moc(=o)r5,其中,n和m为1~2;r4和r5在各情况下独立地为c(12-20)烷基、或c(12-20)链烯基;其中,r3选自氨基烷基、四级氨基烷基、烷氧基烷基、烷氧烷氧基烷基。可离子化脂质分子的例子包括具有式viii表示的结构的化合物。式中,r1和r2为:r1=oc(=o)r4r2=c(=o)zr5,其中,z为nh或氧原子;其中,r4和r5在各情况下独立地为c(12-20)烷基、或c(12-20)链烯基;其中,r3选自:氨基;四级氨基;氨基烷基;四级氨基烷基;nhc(=o)(ch2)pr8;nhc(=o)sr9,其中,r8选自:羧基烷基;氨基烷基;其中,r9选自:氨基烷基;其中,r6各自独立地选自氢原子、烷基、羟基烷基、烷氧基、烷氧烷氧基、氨基烷基;r7选自氢原子、烷基、羟基烷基;q为氧原子或nr7。可离子化脂质分子的例子包括具有式viii-b表示的结构的化合物。式中,r1和r2为:r1=ch2(ch2)noc(=o)r4r2=ch2(ch2)moc(=o)r5其中,n和m为1~2;r4和r5在各情况下独立地为c(12-20)烷基、或c(12-20)链烯基;其中,z为氮原子、氧原子;其中,r3选自:其中,r6各自独立地选自氢原子、烷基、羟基烷基、烷氧基、烷氧烷氧基、氨基烷基;r7选自氢原子、烷基、羟基烷基;p为1~4。可离子化脂质分子的例子包括具有式x表示的结构的化合物。式中,r1和r2为:r1=c(=o)or4r2=nhc(=o)r5其中,r4和r5在各情况下独立地为c(12-20)烷基、或c(12-20)链烯基;其中,r3选自:氨基;四级氨基;氨基烷基;四级氨基烷基;羟基烷基氨基;四级羟基烷基氨基。可离子化脂质分子的例子包括具有式xi表示的结构的化合物。式中,r1和r2为:r1=c(=o)r4r2=c(=o)or5其中,r4和r5在各情况下独立地为c(12-20)烷基、或c(12-20)链烯基;其中,p为1~4;其中,r3选自:其中,r6各自独立地选自氢原子、烷基、羟基烷基、烷氧基、烷氧烷氧基、氨基烷基;r7选自氢原子、烷基;q为氧原子或nr7。结构脂质结构脂质的例子包括胆固醇、甾醇、和类固醇。结构脂质的例子包括胆烷、胆甾烷、麦角甾烷、菜油甾烷(campestane)、多孔甾烷(poriferastane)、豆甾烷、gorgostane、羊毛甾烷、甾烷(gonane)、雌烷、雄烷、孕烷、和环阿屯烷。结构脂质的例子包括甾醇和动物甾醇,例如胆固醇、羊毛甾醇、酵母甾醇、酵母甾烯醇、链甾醇、豆甾烷醇、二氢羊毛甾醇、和7-脱氢胆固醇。结构脂质的例子包括聚乙二醇化胆固醇,和胆甾烷3-氧代-(cl-22)酰基化合物,例如,乙酸胆固醇酯、花生四烯酸胆固醇酯、丁酸胆固醇酯、己酸胆固醇酯、豆蔻酸胆固醇酯、棕榈酸胆固醇酯、山嵛酸胆固醇酯、硬脂酸胆固醇酯、辛酸胆固醇酯、正癸酸胆固醇酯、十二烷酸胆固醇酯、二十四碳烯酸胆固醇酯、壬酸胆固醇酯、正戊酸胆固醇酯、油酸胆固醇酯、反式油酸胆固醇酯、芥酸胆固醇酯、庚酸胆固醇酯、反式亚油酸胆固醇酯、和亚油酸胆固醇酯。结构脂质的例子包括甾醇,例如,植物甾醇、β-谷甾醇、采油甾醇、麦角甾醇、菜籽甾醇、δ-7-豆甾醇、和δ-7-燕麦甾醇。脂质稳定剂脂质稳定剂的例子包括两性离子型脂质。脂质稳定剂的例子包括化合物例如磷脂质。磷脂质的例子包括磷脂酰胆碱、磷脂酰乙醇胺、磷脂酰丝氨酸、磷脂酰肌醇、磷脂酸、棕榈酰油酰基磷脂胆碱、溶血磷脂胆碱、溶血磷脂酰乙醇胺、二棕榈酰磷脂酰胆碱、二油酰磷脂酰胆碱、二硬脂酰磷脂酰胆碱和二亚油酰磷脂酰胆碱。脂质稳定剂的例子包括磷脂酰乙醇胺化合物和磷脂酰胆碱化合物。脂质稳定剂的例子包括l,2-二油酰基-sn-甘油基-3-磷酸胆碱(dopc)。脂质稳定剂的例子包括二植烷酰磷脂酰乙醇胺(dphpe)和l,2-二植烷酰基-sn-甘油基-3-磷酸胆碱(dphpc)。脂质稳定剂的例子包括l,2-二硬脂酰基-sn-甘油基-3-磷酸胆碱(dspc)、l,2-二棕榈酰基-sn-甘油基-3-磷酸胆碱(dppc)、1,2-二棕榈酰基-sn-甘油基-3-磷酸乙醇胺(dppe)、和l,2-二油酰基-sn-甘油基-3-磷酸乙醇胺(dope)。脂质稳定剂的例子包括1,2-二月桂酰基-sn-甘油(dlg)、1,2-二豆蔻酰基-sn-甘油(dmg)、1,2-二棕榈酰基-sn-甘油(dpg)、1,2-二硬脂酰基-sn-甘油(dsg)、l,2-二花生酰基-sn-甘油基-3-磷酸胆碱(dapc)、l,2-二月桂酰基-sn-甘油基-3-磷酸胆碱(dlpc)、l,2-二豆蔻酰基-sn-甘油基-3-磷酸胆碱(dmpc)、l,2-二棕榈酰基-sn-甘油基-0-ethyl-3-磷酸胆碱(dpepc)、l,2-二月桂酰基-sn-甘油基-3-磷酸乙醇胺(dlpe)、1,2-二豆蔻酰基-sn-甘油基-3-磷酸乙醇胺(dmpe)、l,2-二硬脂酰基-sn-甘油基-3-磷酸乙醇胺(dspe)、l-棕榈酰基-2-亚油酰基-sn-甘油基-3-磷酸胆碱、1-棕榈酰基-2-油酰基-sn-甘油基-3-磷酸胆碱(popc)、l-棕榈酰基-2-溶血-sn-甘油基-3-磷酸胆碱(p-lyso-pc);和l-硬脂酰基-2-溶血-sn-甘油基-3-磷酸胆碱(s-lyso-pc)。用于降低免疫原性的脂质用于降低免疫原性的脂质的例子包括聚合物和聚合物-脂质缀合物。用于降低免疫原性的脂质的例子包括具有聚乙二醇(peg)片段的聚乙二醇化脂质。peg片段的分子量可以为任意值。在一些实施方式中,peg片段的分子量可为200、300、350、400、500、550、750、1000、1500、2000、3000、3500、4000或5000da。用于降低免疫原性的脂质的例子包括具有甲氧基聚乙二醇片段的化合物。用于降低免疫原性的脂质的例子包括具有羰基-甲氧基聚乙二醇片段的化合物。用于降低免疫原性的脂质的例子包括具有多支化peg片段的化合物。用于降低免疫原性的脂质的例子包括具有聚甘油片段的化合物。用于降低免疫原性的脂质的例子包括脂质聚合物,例如dspe-mpeg、dmpe-mpeg、dppe-mpeg、和dope-mpeg。用于降低免疫原性的脂质的例子包括peg-磷脂质和peg-神经酰胺。阳离子性脂质阳离子性脂质的例子包括记载于例如us2013/0330401al中的阳离子性hedc化合物。us2013/0115274al公开了一些阳离子性脂质的例子。另外,还有一些阳离子性脂质的例子是本领域已知的。脂质组合物在一些实施方式中,组合物可含有可离子化脂质化合物81、作为结构脂质的胆固醇、作为脂质稳定剂的dopc和dope、和用于降低免疫原性的脂质dppe-mpeg。在某些实施方式中,化合物81可以为组合物的15~25mol%,胆固醇、dopc、和dop的总量可以为组合物的75~85mol%,dppe-mpeg可以为组合物的5mol%。在一个实施方式中,化合物81可以为组合物的25mol%,胆固醇可以为组合物的30mol%,dopc可以为组合物的20mol%,dope可以为组合物的20mol%,dppe-mpeg(2000)可以为组合物的5mol%。纳米粒子本发明的实施方式可提供脂质体纳米粒子组合物。本发明的可离子化分子可用于形成脂质体组合物,所述脂质体组合物可具有脂质类分子的双层结构。纳米粒子组合物可含有一种或多种本发明的可离子化分子,其结构可为脂质体结构、双层结构、胶束、层状结构、或它们的混合物。在一些实施方式中,组合物可包含一种或多种液体介质成分。示于递送本发明的活性剂的液体介质可以是药学上允许的液体介质。液体介质可包含有机溶剂、或水和有机溶剂的组合物。本发明的实施方式可提供大小为10~1000nm的脂质纳米粒子。在一些实施方式中,脂质体纳米粒子的大小可为10~150nm。在某些实施方式中,本发明的脂质体纳米粒子可包封rnai分子,并且,在人血清中暴露1小时后,至少保持80%包封的rnai分子。药物组合物本发明还包括方法将活性剂分配至受试者的器官的方法,所述方法用于通过向受试者施予本发明的组合物来治疗恶性肿瘤。可治疗的器官包括肺、肝脏、胰腺、肾脏、结肠、骨、皮肤、和肠。在一些实施方式中,本发明提供通过向受试者施予本发明的组合物对肺恶性肿瘤疾病进行治疗的方法。在其他方面,本发明提供一系列药物制剂。此处,药物制剂可包含本发明的活性剂、及药物担体、或脂质、以及药学上允许的担体或稀释剂。一般而言,本说明书中的活性剂包括任何用于恶性肿瘤的活性剂,例如,任何抑制核酸分子和任何小分子药物。抑制核酸分子的例子包括核酶、反义核酸、和rna干扰分子(rnai分子)。抗恶性肿瘤药物和抗癌药物的例子包括致癌基因相关因子、肿瘤血管生长因子、转移相关因子、例如tgf-beta等细胞因子、生长因子,例如mmp等蛋白酶、半胱氨酸蛋白酶、和免疫阻遏因子。药物担体能够将组合物导向星状细胞。药物担体可在其内部包含药物,或附着于或含有药物的物质表面,或与药物混合(前提是,所述药物担体包含类视色素衍生物及/或维生素a类似物,并且至少部分暴露于配制物的表面)。对于组合物或配制物而言,可用合适的材料将其包覆,例如,肠溶包衣或随着时间而崩解的材料,或者,将其组入合适的药物释放体系。本发明的药物制剂可含有以下一种或多种成分:表面活性剂、稀释剂、赋形剂、防腐剂、稳定剂、染料、和悬浮剂。用于药物制剂的一些药物担体、稀释剂和成分、以及配置及施予本发明的化合物和组合物的方法记载于remington'spharmaceuticalsciences,18thed.,mackpublishingco.,easton,penn.(1990)。防腐剂的例子包括苯甲酸钠、抗坏血酸,和对羟基苯甲酸酯。表面活性剂的例子包括醇、酯、硫酸化脂肪醇。赋形剂的例子包括蔗糖、葡萄糖、乳糖,淀粉、结晶纤维素、甘露醇、轻质无水硅酸盐、铝酸镁、硅酸镁-铝酸镁复盐(magnesiummetasilicatealuminate)、合成硅酸铝、碳酸钙、碳酸氢钠、磷酸氢钙、和羧甲基纤维素钙。悬浮剂的例子包括椰子油、橄榄油、芝麻油、花生油、豆油(soya)、邻苯二甲酸乙酸纤维素,乙酸甲酯-甲基丙烯酸酯共聚物、和邻苯二甲酸酯。用于递送一种或多种具有基因沉默活性的分子的本发明的治疗制剂可施予至需要的哺乳动物。可向哺乳动物以包封于脂质体的形式施予治疗有效量的制剂和活性剂,由此对恶性肿瘤进行预防或治疗。施予途径可以为局部或全身。本发明的具有治疗效果的制剂可通过各种途径施予,例如,静脉、腹腔内、肌肉内、皮下、和口服。施予途径可包括胃肠外递送,例如,肌肉内、皮下、静脉、髓内注射,以及鞘内、直接心室内、腹腔内,鼻内、或眼内注射。制剂还可以以持续释放剂型或控释剂型施予,包括长效注射、渗透泵等,由此以事先规定的速度进行长期及/或定时、脉冲式的施予。本发明的组合物可通过各种途径施予,包括,口服途径和胃肠外途径,作为例子,包括但不限于:口服、静脉、肌肉内、皮下、局部、肺内、气道内、气管内、支气管内、鼻、直肠、动脉内、门脉内、心室内、髓内、淋巴结内、淋巴内、脑内、鞘内、脑室内、经粘膜、经皮、鼻内、腹腔内、和子宫内途径,并且,本发明的组合物可配制成适合各种施予途径的剂型。上述剂型和配制方法可从任何已知的剂型和方法中适当选择。例如,参见hyojunyakuzaigaku,standardpharmaceutics,ed.byyoshiteruwatanabeetal.,nankodo,2003。适合口服施予的剂型的例子包括但不限于粉剂、颗粒、片剂、胶囊、液体、悬浮液、乳剂、凝胶、糖浆,适合胃肠外施予的剂型的例子包括注射剂,例如,可注射的溶液、可注射的悬浮液、可注射的乳剂、和准备用于注射的制剂。用于胃肠外施予的制剂可为水性或非水性的等渗无菌溶液或悬浮液。用于胃肠外施予、例如弹丸注射(bolusinjection)或持续输液的药物制剂包括以可溶于水的形式含有活性制剂的水溶液。活性化合物的悬浮液可制备成合适的油性注射悬浮液。水性注射悬浮液可含有提高悬浮液粘性的物质,例如羧甲基纤维素钠、山梨糖醇、或葡聚糖。另外,为了制备高浓度的溶液,悬浮液可选择性地含有合适的稳定剂或提高化合物溶解性的成分。用于注射的制剂可以单位剂型的形式存在,例如,可与添加的防腐剂共存于安瓿或多剂量容器中。制剂可采用例如利用油性或水性介质的悬浮液、溶液或乳液,并可含有例如悬浮剂、稳定剂及/或分配剂等配制用剂。另外,活性成分可以为粉剂形式,并在使用前与合适的介质、例如无菌无热原水混合。除了前文记载的配制物以外,制剂还可被配置成贮库(depot)制剂。这种长效制剂可通过肌肉注射施予。因此,例如,制剂可使用合适的聚合物材料或疏水性材料配制,例如,用允许的油配制的乳剂、或离子交换树脂、或例如难溶盐等难溶衍生物。本发明的组合物和制剂还可用于局部递送,并可通过任何使用局部递送介质的步骤而应用于受试者的皮肤。例如,可手动使用试剂,也可采用涂敷器(applicator)、或包含二者的步骤。敷用之后,可通过例如摩擦而使制剂渗入受试者的皮肤。敷用可每天实施多次,也可每天实施1次。例如,制剂敷用于受试者皮肤的频率可为每天1次、每天2次、或每天多次,或者,每2天1次、每3天1次、或约每周1次、每2周1次、或数周1次。本文中记载的制剂或药物组合物可通过任何合适的手段施予至受试者。施予方法的例子包括:(a)通过注射(皮下、腹腔内、静脉、肌肉内、皮内、眼眶内、囊内、脊柱内、胸骨等),包括输液泵递送;(b)局部施予,例如,通过例如长效植入而直接注入肾脏或心区,以及本领域技术人员认可的使活性化合物与活体组织接触的合适手段;等等。对于药物组合物而言,具体的制剂、施予途径和用量可以由医师根据患者的情况选择。例如,参见goodman&gilman'sthepharmacologicalbasisoftherapeutics,12thed.,sec.1,2011。典型地,向患者施予的组合物用量的范围可以是为0.5~1000mg/kg(患者体重)。根据患者的需要,可在1天或数天期间单次、或连续2次或多次施予上述用量。已经确立了至少某些病症的化合物的人用剂量的情况下,用量可与已经确定的人用剂量大致相等,或为所述剂量的0.1%~500%左右,更优选为25%~250%左右。尚未确立人用剂量,即作为新发现的药物组合物的情况下,可根据ed50或id50的值、或其他通过体外或体内研究而得到、且经过使用动物的毒性试验、效果试验验证的合适的值来推测合适的人用剂量。预防或治疗恶性肿瘤的方法本发明还涉及用于控制恶性肿瘤的活跃度或生长的方法,所述方法包括向有需要的受试者施予有效量的组合物。此处,所谓有效量,是指在治疗恶性肿瘤的方法中,能够缓解症状、或者延迟或终止肿瘤进展的量,更优选为能够预防恶性肿瘤的发病或复发、或者治愈恶性肿瘤的量。另外,也优选使施予利大于弊的量。这样的剂量可通过使用培养细胞的体外试验、或者模型动物或例如小鼠、大鼠、狗、猪等哺乳动物的试验适当确定,并且,可使用本领域技术人员熟知的试验方法。另外,担体中的活性剂量和本发明的方法中使用的活性剂量可以是本领域技术人员已知的剂量,或可通过上述试验而适当确定。施予频率取决于使用的组合物的性能和上述受试者的病症,可以为每天数次(即,每天2、3、4、5、次或更多)、每天1次、数天1次(即,每2、3、4、5、6或7天等)、每周1次(例如,每周2、3、4次等),每2周数次、或每数周1次(例如,每2、3、4周等)。在一些实施方式中,本发明还涉及利用上述担体将药物递送至恶性肿瘤细胞的方法。该方法包括下述步骤:向活体或介质(例如,含有生产细胞外基质的肺细胞的培养基)施予或添加载有应递送物质的担体。这些步骤可按照任何已知的方法或本发明中记载的方法适当地完成。另外,上述方法包括在体外实施的模式、和以体内的肺部恶性肿瘤细胞作为靶标的模式。可将本发明的具有治疗效果的制剂全身性地递送施予,由此使活性剂广泛地分配于体内。本发明的实施方式可提供包含本发明的治疗性分子、和药学上允许的担体的治疗制剂。对于本发明的制剂的有效量而言,可每天施予1~12次,或每周施予1次。施予期间可以为1、2、3、4、5、6或7天,或者1、2、3、4、5、6、8、10或12周。其他实施方式用于分配治疗受试者的恶性肿瘤的活性剂的组合物,所述组合物含有可离子化脂质、结构脂质、和用于降低纳米粒子的免疫原性的脂质。恶性肿瘤可位于肺、结肠、肾脏或胰腺。恶性肿瘤可位于肝脏、骨、皮肤、或肠。可离子化脂质可以为组合物中的脂质的50mol%~80mol%,或组合物中的脂质的55mol%~65mol%。结构脂质可以为组合物中的脂质的20mol%~50mol%,或组合物中的脂质的35mol%~55mol%。结构脂质可以为胆固醇、甾醇、或类固醇。用于降低纳米粒子的免疫原性的脂质可以为组合物中的脂质的1mol%~8mol%。用于降低纳米粒子的免疫原性的脂质可以具有聚乙二醇(peg)片段,所述聚乙二醇片段的分子量为200~5000da。用于降低纳米粒子的免疫原性的脂质可以为dppe-mpeg、dspe-mpeg、dmpe-mpeg、或dope-mpeg。本发明还包括用于分配治疗受试者的恶性肿瘤的活性剂的组合物,所述组合物含有可离子化脂质、结构脂质、一种或多种脂质稳定剂、和用于降低纳米粒子的免疫原性的脂质。可离子化脂质可以为组合物中的脂质的15mol%~40mol,或组合物中的脂质的20mol%~35mol%。结构脂质可以为组合物中的脂质的25mol%~40mol%,或组合物中的脂质的30mol%~35mol%。一种或多种脂质稳定剂的总量可以为组合物中的脂质的25mol%~40mol%,其中,各脂质稳定剂可各自为5mol%~35%mol%。一种或多种脂质稳定剂可以为磷脂酰乙醇胺化合物或磷脂酰胆碱化合物。组合物(化合物81/胆固醇/dopc/dope/dppe-peg-2000)可以为下述中的1种组合,其中,数字表示成分的mol%浓度:(25/30/30/10/5)、(25/30/25/15/5)、(25/30/20/20/5)、(25/30/15/25/5)、(25/30/10/30/5)、(25/35/15/20/5)、(25/35/20/15/5)、(30/30/15/20/5)、(30/30/20/15/5)、(35/30/15/15/5)。在一些实施方式中,可离子化脂质可以是化合物81,结构脂质可以是胆固醇,脂质稳定剂可以是dopc和dope,用于降低纳米粒子的免疫原性的脂质可以是dspe-mpeg-2000,其中,化合物81占组合物的15~25mol%,胆固醇、dopc、和dope共同占组合物的75~85mol%,dspe-mpeg-2000占组合物的1~5mol%,化合物81、胆固醇、dopc、dope、和dspe-mpeg-2000实质上共同占组合物中的脂质的100mol%。活性剂可以是以gst-π作为靶标的rnai分子和以p21作为靶标的rnai分子。在一些实施方式中,活性剂包含以gst-π作为靶标的rnai分子和以p21作为靶标的rnai分子,组合物包含包封rnai分子的脂质体纳米粒子。在某些实施方式中,活性剂包含以gst-π作为靶标的rnai分子和以p21作为靶标的rnai分子,组合物包含包封rnai分子的脂质体纳米粒子,并且,在人血清中暴露1小时后,所述脂质体纳米粒子保持至少80%的包封的rnai分子。本发明还包括含有脂质组合物和活性剂的药物组合物。活性剂可以是一种或多种rnai分子。用于治疗恶性肿瘤的rnai分子包括gst-π作为靶标的rnai分子和以p21作为靶标的rnai分子。用于治疗恶性肿瘤的rnai分子可为例如sirna、shrna、或micro-rna,以及dna指导的rna(ddrna)、piwi-相互作用rna(pirna)、和重复相关sirna(rasirna)。在某些实施方式中,本发明提供含有用于治疗恶性肿瘤的rnai分子的药物组合物,所述rnai分子仅以gst-π作为靶标。本发明的实施方式还包括将活性剂分配至受试者的器官的方法,所述方法通过向受试者施予本发明的组合物来治疗恶性肿瘤。恶性肿瘤可位于肺、结肠、肾脏或胰腺。恶性肿瘤可位于肝脏、骨、皮肤、或肠。恶性肿瘤可位于选自由头、颈部、脑、乳房、食道、胃、肠、十二指肠、肝脏、胆囊、胆管、肾脏、尿道、膀胱、前列腺、睾丸、子宫、卵巢、皮肤、骨、骨髓、血液、皮肤、和上皮层组成的组中的任何解剖学部位。对于施予而言,可以以0.01~2mg/kg的量施予rnai分子,每天至少施予1次,持续期间为12周以下。施予可提供的gst-πrnai分子的平均auc(o-last)为1~1000ug*min/ml,平均cmax为0.1~50ug/ml。施予可提供的p21rnai分子平均血浆auc(o-last)为1~1000ug*min/ml,平均血浆cmax为0.1~50ug/ml。本发明提供对需要的哺乳动物、或任何动物的恶性肿瘤的一种或多种症状进行预防、治疗或改善的方法,所述方法包括向哺乳动物施予治疗有效量的含有rnai分子的组合物,其中,部分rnai分子具有降低gst-π表达的活性,部分rnai分子具有降低p21表达的活性。在一些实施方式中,恶性肿瘤与哺乳动物中的kras突变相关,上述方法还包括对哺乳动物中的肿瘤细胞进行鉴定,所述肿瘤细胞具有以下至少一者:(i)kras基因突变、和(ii)kras蛋白表达水平异常。本发明的方法可应用于包括人在内的任何动物。哺乳动物可以为人,gst-π可以为人gst-π,p21可以为人p21。在一些实施方式中,恶性肿瘤过量表达gst-π,若gst-π的表达被下调,则p21水平可能也会被下调。rnai分子能够降低哺乳动物中的gst-π和p21表达。施予使哺乳动物中的gst-π和p21表达降低至少5%、且持续至少5天。本发明的方法可减轻恶性肿瘤的一种或多种症状,或者延迟或终止恶性肿瘤的发展,或减缓受试者中恶性肿瘤细胞的生长。较之正常细胞而言,肿瘤细胞可含有以更高水平表达的野生型kras蛋白。肿瘤细胞可过量表达野生型gst-πrna或蛋白,若gst-π的表达被阻遏,则p21rna或蛋白的水平可能被提高。在一些实施方式中,肿瘤细胞可在kras蛋白的第12、13和第61位残基具有突变。在某些实施方式中,肿瘤细胞可含有kras蛋白突变,肿瘤为选自肠癌、胰腺癌和肺癌中的癌症。肿瘤可以选自由肺腺癌、粘液腺癌、胰腺导管癌、和结肠直肠癌组成的组。恶性肿瘤可以是选自由肺腺癌、粘液腺癌、胰腺导管癌、结肠直肠癌、乳腺癌、和纤维肉瘤组成的组中的肿瘤。恶性肿瘤可位于任何解剖学部位,在一些实施方式中,可以是选自由肺、肝脏、胰腺、结肠、肾脏、骨、皮肤、肠、和它们的任意的组合组成的组。另一方面,本发明涉及以下出乎意料的发现:通过利用gst-π和p21的sirna抑制剂进行治疗,能够使体内恶性肿瘤的大小缩小。另外,本发明还获得了出乎意料的有利结果,即,肿瘤细胞凋亡可被提高至协同致死(syntheticlethality)的水平,由此,可提供用于预防或治疗恶性肿瘤的方法和组合物。本发明涉及的方法和组合物结合了基于核酸的治疗性化合物,其用于递送至各种器官,由此对恶性肿瘤的疾病及病症进行预防、治疗、或改善。在一些实施方式中,本发明提供rna干扰分子(rnai分子)的组合物,用于使各种与恶性肿瘤有关的靶基因沉默。本发明可提供用于递送治疗分子的组合物、及使用其的方法。本发明的各种基于rna的药物和组合物可用于预防或治疗恶性肿瘤的方法。在一些实施方式中,通过利用调节gst-π和p21的表达水平的sirna剂进行治疗,可使具有kras突变、或呈现异常的kras表达水平的恶性肿瘤缩小至能够引起肿瘤细胞的协同致死的水平。本发明涉及针对恶性肿瘤的、基于核酸的治疗性化合物的方法和组合物。在一些实施方式中,本发明提供能够使gst-π和p21的表达沉默的rnai分子、结构和组合物。本申请公开的结构和组合物可用于恶性肿瘤的预防、治疗或使其大小缩小。本发明提供可用于治疗受试者中的肿瘤的组合物和方法。具体而言,本发明提供能使gst-π核酸分子或多肽、及p21核酸分子或多肽的表达降低的治疗性组合物,由此对与kras相关的肿瘤进行治疗。在一些方面,本发明包括与gst-π核酸分子的至少1个片段对应或互补的抑制核酸分子,并且,所述抑制核酸分子使gst-π在细胞中的表达降低。在其他方面,本发明包括与p21核酸分子的至少1个片段对应或互补的抑制核酸分子,所述抑制核酸分子使p21在细胞中的表达降低。在某些实施方式中,本发明提供以下用于阻遏gst-π和p21的双链核酸分子:例如sirna或shrna等rnai分子、dna指导的rna(ddrna)、piwi-相互作用rna(pirna)、和重复相关sirna(rasirna)。本发明的方法可应用于包括人在内的任何动物。在一些方面,本发明包括一种或多种编码上述抑制核酸分子的载体。载体可以是逆转录病毒载体、腺病毒载体、腺相关病毒载体、或慢病毒载体。在另一些实施方式中,载体可含有适于哺乳动物细胞中表达的启动子。其他的实施方式包括具有kras突变或呈现异常的kras表达水平的癌细胞,并且,可还包括载体、或上述的任一种抑制核酸分子。在另一些实施方式中,细胞可以是体内肿瘤细胞。在一些实施方式中,本发明包括使gst-π和p21在恶性肿瘤细胞中的表达降低的方法,其中,所述恶性肿瘤细胞具有kras突变或呈现异常的kras表达水平。所述方法可包含使细胞与有效量的抑制核酸分子接触,其中,抑制核酸分子抑制gst-π多肽和p21多肽的表达,从而使gst-π和p21在细胞中的表达降低。在另一些实施方式中,本发明的方法能够使gst-π和p21在恶性肿瘤的转录或翻译水平降低。在某些实施方式中,本发明包括使gst-π和p21在恶性肿瘤细胞中的表达降低的方法,其中,所述细胞可以是人细胞、肿瘤细胞、体内细胞、或体外细胞。本发明的实施方式还可提供治疗患有肿瘤的受试者的方法,其中,癌肿瘤细具有kras突变或呈现异常的kras表达水平。该方法可包括向受试者施予有效量的2种或多种抑制核酸分子,其中,抑制核酸分子使gst-π和p21的表达降低,由此对肿瘤进行治疗。在一些实施方式中,本发明的方法可使肿瘤的大小小于治疗之前或未经治疗的肿瘤。在各种实施方式中,可将抑制核酸分子载于脂质体、聚合物、微球、纳米颗粒,基因治疗载体、或裸dna载体而进行递送。在其他方面,本发明的特征在于,在对受试者(例如,患有肿瘤的病人)进行治疗的方法中,肿瘤癌细胞具有kras突变或呈现异常的kras表达水平。在某些实施方式中,方法可包括向受试者施予有效量的抑制核酸分子,其中,所述抑制核酸分子为具有rna干扰活性的反义核酸分子、sirna、dsrna,或它们的组合,并且,能够抑制gst-π多肽和p21多肽的表达。在某些实施方式中,肿瘤细胞过量表达gst-π,若gst-π的表达被阻遏,则p21rna或蛋白的水平可能被提高。在某些实施方式中,肿瘤可以是恶性肿瘤、肺癌、肾癌或胰腺癌。脂质尾部的结构本发明的脂质类化合物可具有一个或多个亲脂性尾(lipophilictails),其含有一个或多个烷基或链烯基。亲脂性尾的例子包括c(14:l(5))链烯基、c(14:l(9))链烯基、c(16:l(7))链烯基、c(16:l(9))链烯基、c(18:1(3))链烯基、c(18:l(5))链烯基、c(18:l(7))链烯基、c(18:l(9))链烯基、c(18:l(11))链烯基、c(18:l(12))链烯基、c(18:2(9,12))链烯基、c(18:2(9,11))链烯基、c(18:3(9,12,15))链烯基、c(18:3(6,9,12))链烯基、c(18:3(9,11,13))链烯基、c(18:4(6,9,12,15))链烯基,c(l8:4(9,11,13,15))链烯基、c(20:1(9))链烯基、c(20:l(11))链烯基、c(20:2(8,11))链烯基、c(20:2(5,8))链烯基、c(20:2(11,14))链烯基、c(20:3(5,8,11))链烯基、c(20:4(5,8,11,14))链烯基、c(20:4(7,10,13,16))链烯基、c(20:5(5,8,11,14,17))链烯基、c(20:6(4,7,10,13,16,19))链烯基、c(22:l(9))链烯基、c(22:l(13))链烯基、和c(24:l(9))链烯基。化学定义本文中,用语“烷基”表示作为饱和脂肪族基团的烃基,其可以为任何长度。烷基可以是支链或直链、经取代或未取代的含有1~22个碳原子的脂肪族基团。该定义同样适用于例如环烷基、烷氧基、酰基、和芳烷基等其他基团的烷基部分。本文中,例如,用语“c(l-5)烷基”包括c(l)烷基、c(2)烷基、c(3)烷基、c(4)烷基、和c(5)烷基。同样地,例如,用语“c(3-22)烷基”包括c(l)烷基、c(2)烷基、c(3)烷基、c(4)烷基、c(5)烷基、c(6)烷基、c(7)烷基、c(8)烷基、c(9)烷基、c(10)烷基、c(ll)烷基、c(12)烷基、c(13)烷基、c(14)烷基、c(15)烷基、c(16)烷基、c(17)烷基、c(18)烷基、c(19)烷基、c(20)烷基、c(21)烷基、和c(22)烷基。本文中,可利用以下用语指定烷基:例如,me(甲基)、et(乙基)、pr(任何丙基)、npr(n-pr、正丙基),'pr(i-pr、异丙基),bu(任何丁基)、nbu(n-bu、正丁基),3/4u(i-bu,异丁基)、3/4u(s-bu、仲丁基),和*bu(t-bu,叔丁基)。本文中,用语“链烯基”表示具有至少一个碳-碳双键的烃基。链烯基可以是支链或直链、经取代或未取代的含有2~22个碳原子和至少1个碳-碳双键的烃基。本文中,用语“取代”表示一个或多个取代的原子,或者可以相同或不同的取代基,其中,取代基包括氢。因此,例如,用语烷基、环烷基、链烯基、烷氧基、酰基、芳烷基等用语所表示的基团可包含经取代的变体。取代变体包括线性、支链、和环状变体,以及以一个或多个取代基对与基团中的任意的碳原子结合的一个或多个氢原子进行取代而得到的基团。一般而言,化合物可具有一个或多个手性中心。具有一个或多个手性中心的化合物可包括被称为“异构体”、“立体异构体”、“非对映异构体”、“对映异构体”、“光学异构体”、“外消旋混合物”的化合物。立体化学命名的惯例、例如立体异构体命名的cahn-ingold-prelog规则、以及立体化学测定、立体异构体区分的方法均是本领域已知的。例如,参见michaelb.smithandjerrymarch,march’sadvancedorganicchemistry,5thedition,2001。本申请公开的化合物和结构应包含所有针对特定的化合物或结构理应存在的异构体、立体异构体、非对映异构体、对映异构体、及/或光学异构体,并且包括它们的任何混合物、外消旋体或其他。本发明包括本文中公开的化合物和组合物的任何及所有互变异构体形式、溶剂合物或非溶剂化形式、水合物或未水合形式、及任何原子同位素形式。本发明包括本文中公开的化合物和组合物的任何及所有多晶型或不同的晶型。体外敲低的方案例在转染前1天,向96孔板中注入细胞(2x103细胞/孔,100μl含有10%fbs的dmem(hyclonecat.#sh30243.01)),在37℃的孵育器(湿润气氛,空气中co2浓度为5%)中培养。转染前,将培养基置换为90μl含有2%fbs的opti-memi减血清培养基(lifetechnologiescat.#31985-070)。然后,将0.2μl的lipofectaminernaimax(lifetechnologiescat.#13778-100)与4.8μl的opti-memi于室温混合5分钟。接着,将1μl的sirna与4μl的opti-memi混合,进而加入lf2000溶液,轻轻混合,无需振荡(vortex)。于室温反应5分钟,然后,将混合物于室温进一步孵育10分钟,形成rna-rnaimax复合物。接下来,向每个孔中加入10μl的rna-rnaimax复合物,用手轻轻摇动平板。将细胞在37℃的孵育器(湿润气氛,空气中co2浓度为5%)中培养2小时。将培养基置换为新鲜的含有2%fbs的opti-memi减血清培养基。转染24小时后,用冰冷的pbs洗涤1次细胞。用50μl的cell-to-ct裂解液(lifetechnologiescat.#4391851c)裂解细胞,于室温处理5-30分钟。加入5μl的终止溶液,并于室温孵育2分钟。立刻使用taqman通过rt-qpcr测定mrna水平。也可将试样于-80℃冷冻,在稍后进行分析。血清稳定性的方案例将0.2mg/ml的sirna和10%人血清一同于37℃孵育。在特定的时间点(0、5、15和30分钟),用200μl提取溶剂(氯仿:苯酚:异戊醇=24:25:1)等份地提取200μl的试样。振荡试样,并于13,000rpm、室温离心分离10分钟,然后转移上层溶液,用0.45μm过滤器进行过滤。将滤液移至300μl的hplc注射瓶中。对于lcm而言,流动相为mpa:100mmhfip+7mmtea(水中),mpb:50%甲醇+50%乙腈。柱:watersacquityost2.1x50mm,1.7μm。实施例实施例1:本发明的gst-π和p21的sirna制剂使癌异种移植肿瘤在体内显著缩小。在以脂质体制剂的形式施予至癌异种移植肿瘤的情况下,gst-π和p21的sirna在体内发挥了基因敲低效能。图1显示了gst-π(序列号61、126)和p21(序列号341、355,n=u)sirna的脂质体制剂的肿瘤抑制效果。利用的癌异种移植模型被施予了相对较低的量,p21sirna为0.74mg/kg。gst-π和p21的sirna制剂在施予后数天内呈现了显著且出乎意料的有利的肿瘤抑制效果。30天后,gst-π和p21的sirna呈现了明显有利的肿瘤抑制效果,较之对照而言,肿瘤体积缩小超过2倍。gst-πsirna以包含组合物(可离子化脂质:胆固醇:dope:dopc:dppe-peg-2k)(25:30:20:20:5)的脂质体制剂的形式注射施予了4次(第1、8、15、22天)。对于癌异种移植模型而言,从atcc获得了a549细胞系。在添加有10%胎牛血清、100u/ml盘尼西林和100μg/ml链霉素的培养基中维持细胞。在接种前48小时稀释(split)细胞,从而使细胞在被收集时处于对数生长期。使用胰蛋白酶-edta轻度消化细胞后,从组织培养物中收集细胞。在台盼蓝的存在下,用血细胞计数器计数并测定活细胞的数量(仅计数活细胞)。在无血清培养基中重新悬浮细胞,使浓度为5x107/ml。然后将细胞悬浮液与冰上融化的bdmatrigel以1:1的比例充分混合,用于注射。小鼠为charlesriverlaboratory的雌性无胸腺裸鼠(nu/nu),经过免疫妥协,6~8周龄,每组7~8只小鼠。对于肿瘤模型的制备而言,使用25g注射针和针筒,在每只小鼠的右侧腹皮下接种0.1ml含有2.5x106个a549细胞的接种液,每只小鼠接种1剂接种液。小鼠在接种时未被麻醉。对于肿瘤体积的测量和随机化而言,肿瘤大小的测量结果取至0.1mm。肿瘤体积利用下式算出:肿瘤体积=长x宽2/2。当培养的肿瘤生长至约120~175mm3、平均肿瘤体积为150mm3时,将小鼠分配至各介质对照组和处理组,结果,处理组的平均肿瘤体积为介质对照组的平均肿瘤体积的10%以下,符合理想地,肿瘤体积的cv%小于25%。按照给药方案,在同一天施予试验制剂和对照介质。在第1周检测3次肿瘤体积,在接下来的数周,每周检测2次,包括研究结束当天。对于施予量而言,在施予当天,从-80℃冰箱中取出试验制剂,在冰上融化。在装入注射器之前,手动将装在瓶中的制剂颠倒数次。所有试验制剂均以10ml/kg通过静脉注射施予。对于体重而言,小鼠称重至0.1g。在第1次施予后的7天内,每天检测并记录体重。在接下来的数周,每周检测并记录体重2次,包括研究结束当天。对于肿瘤的采集而言,在第1次施予后28天,测量肿瘤体积,切割肿瘤供于称重,并保存以用于pd生物标志物研究。记录肿瘤的重量。实施例2:本发明的gst-π和p21的sirna制剂使癌异种移植肿瘤在体内显著缩小。在以脂质体制剂的形式施予至癌异种移植肿瘤的情况下,gst-π和p21的sirna在体内发挥了基因敲低效能。图2显示了gst-π(序列号156、182)和p21(序列号341、355,n=u)sirna的脂质体制剂的肿瘤抑制效果。利用的癌异种移植模型被施予了相对较低的量,各sirna为0.75mg/kg。gst-π和p21的sirna制剂在施予后数天内呈现了显著且出乎意料的有利的肿瘤抑制效果。30天后,gst-π和p21的sirna呈现了明显有利的肿瘤抑制效果,较之对照而言,肿瘤体积缩小1.7倍。实施例3:本发明的gst-π和p21的sirna制剂在体外展示出由癌细胞凋亡导致的癌细胞死亡上升。体外癌细胞凋亡的监控通过观察puma的表达上调进行,puma是细胞凋亡的生物标志物,其与细胞活性的下降相关。gst-π和p21的sirna制剂出乎意料地使癌细胞的凋亡上升。如图3所示,在gst-π和p21的sirna转染后2~4天,gst-π和p21的sirna制剂使puma的表达水平显著升高(图3,p21(a)+gstp(a),p21:序列号341、355,n=u,gstp:序列号156、182)。上述数据表明,本发明的gst-π和p21的sirna制剂导致出乎意料的癌细胞凋亡的上升。puma生物标志物的方案如下。在转染前1天,向96孔板中注入细胞(2x103细胞/孔,100μl含有10%fbs的dmem(hyclonecat.#sh30243.01)),以2x103细胞/孔注入96孔板,在37℃的孵育器(湿润气氛,空气中co2浓度为5%)中培养。第二天,在转染前将培养基置换为90μl含有2%fbs的opti-memi减血清培养基(lifetechnologiescat.#31985-070)。然后,将0.2μl的lipofectaminernaimax(lifetechnologiescat.#13778-100)与4.8μl的opti-memi于室温混合5分钟。将1μl的sirna(储存浓度:1μμ)与4μl的opti-memi混合,加入rnaimax溶液,轻轻混合。将混合物于室温孵育10分钟,形成rna-rnaimax复合物。向每个孔中加入10μl的rna-rnaimax复合物,使sirna最终浓度为10nm。将细胞孵育2小时,并将培养基置换为新鲜的含有2%fbs的opti-memi减血清培养基。在转染后1、2、3、4、6天,用冰冷的pbs洗涤1次细胞,然后用50μl的cell-to-ct裂解液(lifetechnologiescat.#4391851c)于室温裂解5~30分钟。加入5μl的终止溶液,并于室温孵育2分钟。使用taqman通过rt-qpcr测定puma(bbc3,cat#hs00248075,lifetechnologies)mrna水平。实施例4:以gst-π和p21作为靶标的sirna的双敲低体外研究表明,上述组合能够高效地阻遏2种蛋白质的mrna。如表11所示,浓度为2、10、50nm时,各gst-π和p21敲低水平高。表11:a549中的双敲低方案:用2、10、50nm的p21sirna进行转染。在1、2、3、或4天后用10nm的gst-πsirna进行转染。1天后,通过rt-pcr分析p21mrna和gst-πmrna的水平。实施例5:原位a549肺癌小鼠模型。本发明的gst-πsirna能够导致原位肺癌肿瘤在体内显著缩小。该实施例中,在以脂质体制剂的形式施予至无胸腺裸鼠的原位肺癌肿瘤的情况下,gst-πsirna在体内发挥了基因敲低效能。一般而言,原位肿瘤模型可呈现药物效果、效能和提高的预估性能的直观临床意义。原位肿瘤模型中,肿瘤细胞被直接植入与细胞来源种类相同的器官。将处理组和介质对照组剖检时的最终原发瘤重量进行比较,由此对sirna制剂对于人a549肺癌的抗肿瘤效果进行评价。图4显示了由基于bu2结构的gst-πsirna(序列号61、126)导致的体内原位肺癌肿瘤抑制。向使用的原位a549肺癌小鼠模型以相对较低的量(2mg/kg)施予以gst-π为靶标的sirna。在为期6周的该研究中,gst-πsirna呈现了显著且出乎意料的有利的肿瘤抑制效果。如图4所示,43天后,gst-πsirna呈现了明显有利的肿瘤抑制效果,较之对照而言,最终肿瘤平均重量大幅度减轻2.8倍。该研究中,使用了5~6周龄的雄性ncrnu/nu小鼠。在实验期间,将实验动物饲育在经过fipea过滤的环境中。sirna制剂在使用前于4℃保存,并在注射进小鼠10分钟前使其升温至室温。对于该a549人肺癌原位模型而言,在外科原位移植(soi)当天,从接受a549肿瘤异种移植的动物皮下部位采集备份肿瘤(stocktumor),置于rpmi-1640培养基中。摘除坏死组织,将活体组织切割成1.5~2mm3的小片。通过异氟烷吸入将动物麻醉,用碘酒和酒精消毒手术区。使用一对手术剪在小鼠左胸壁切开约1.5cm长的横向切口。切开第三、四肋骨之间,暴露左肺。使用8-0外科缝线(尼龙),将一片a549肿瘤移植至肺表面。使用6-0缝线(丝)将胸壁缝合。使用装有25gx11/2针头的3cc注射器进行胸内穿刺,使肺重新膨胀,由此排出胸腔中残余的空气。使用6-0缝线(丝)将胸壁缝合。上述所有手术步骤均在hepa过滤层流操作台上使用7x放大倍数的显微镜实施。肿瘤移植3天后,将荷瘤模型小鼠随机分组,每组10只小鼠。对于处理组而言,在肿瘤移植3天后对10只小鼠进行处理。对于处理组而言,制剂为脂质体组合物(可离子化脂质:胆固醇:dope:dopc:dppe-peg-2k:dspe-peg-2k)。脂质体包封gst-πsirna。对于研究终点而言,在处理开始42天后杀死实验小鼠。切除原发瘤,并用电子天平称重,用于后续分析。对于化合物毒性的估测而言,在整个实验期间,处理组和对照组的小鼠的平均体重维持在正常范围内。在小鼠中未观察到其他毒性症状。实施例6:本发明的gst-π和p21的sirna制剂使癌异种移植肿瘤在体内显著缩小。在以脂质体制剂的形式施予至癌异种移植肿瘤的情况下,gst-π和p21的sirna在体内发挥了基因敲低效能。图5显示了gst-πsirna(序列号:156、182)的体内肿瘤抑制效果。利用的癌异种移植模型被施予了相对较低的量(以gst-π作为靶标的sirna为0.75mg/kg)。gst-π的sirna制剂在施予后数天内呈现了显著且出乎意料的有利的肿瘤抑制效果。36天后,gst-πsirna呈现了明显有利的肿瘤抑制效果,较之对照而言,肿瘤体积2倍。如图5所示,gst-πsirna在研究结束当天展示出显著且出乎意料的有利的肿瘤抑制效果。具体而言,肿瘤重量减轻超过2倍。gst-πsirna以包含组合物(可离子化脂质:胆固醇:dope:dopc:dppe-peg-2k)(25:30:20:20:5)的脂质体制剂的形式注射施予了2次(第1、15天)。对于癌异种移植模型而言,从atcc获得了a549细胞系。在添加有10%胎牛血清、100u/ml盘尼西林和100μg/ml链霉素的培养基中维持细胞。在接种前48小时稀释细胞,从而使细胞在被收集时处于对数生长期。使用胰蛋白酶-edta轻度消化细胞后,从组织培养物中收集细胞。在台盼蓝的存在下,用血细胞计数器计数并测定活细胞的数量(仅计数活细胞)。在无血清培养基中重新悬浮细胞,使浓度为5x107/ml。然后,将细胞悬浮液与冰上融化的bdmatrigel以1:1的比例充分混合,用于注射。小鼠为charlesriverlaboratory的雌性无胸腺裸鼠(nu/nu),经过免疫妥协,6~8周龄,每组7~8只小鼠。对于肿瘤模型的制备而言,使用25g注射针和针筒,在每只小鼠的右侧腹皮下接种0.1ml含有2.5x106个a549细胞的接种液,每只小鼠接种1剂接种液。小鼠在接种时未被麻醉。对于肿瘤体积的测量和随机化而言,肿瘤大小的测量结果取至0.1mm。肿瘤体积利用下式算出:肿瘤体积=长x宽2/2。当培养的肿瘤生长至约120~175mm3、平均肿瘤体积为150mm3时,将小鼠分配至各介质对照组和处理组,结果,处理组的平均肿瘤体积为介质对照组的平均肿瘤体积的10%以下,符合理想地,肿瘤体积的cv%小于25%。按照给药方案,在同一天施予试验制剂和对照介质。在第1周检测3次肿瘤体积,在接下来的数周,每周检测2次,包括研究结束当天。对于施予量而言,在施予当天,从-80℃冰箱中取出试验制剂,在冰上融化。在装入注射器之前,手动将装在瓶中的制剂颠倒数次。所有试验制剂均以0.75mg/kg通过静脉注射施予,每2周1次(q2w)x2,10ml/kg。对于体重而言,小鼠称重至0.1g。在第1次施予后的7天内,每天检测并记录体重。在接下来的数周,每周检测并记录体重2次,包括研究结束当天。对于肿瘤的采集而言,在第1次施予后28天,测量肿瘤体积,切割肿瘤供于称重,并保存以用于pd生物标志物研究。记录肿瘤的重量。实施例7:本发明的gst-πsirna制剂在体外展示出由癌细胞凋亡导致的癌细胞死亡上升。gst-πsirna发挥了gst-π敲低效果,由此导致与细胞活性下降相关的细胞凋亡生物标志物puma的上调。对于序列号156、182的gst-πsirna而言,其含有位于种子区域的脱氧核苷酸组合、2’-氟代脱氧核苷酸、和2’-ome取代核糖核苷酸,并出乎意料地使癌细胞的凋亡上升。如图6所示,对由序列号156、182的gst-πsirna导致的puma的表达水平进行了测量。图6中,在gst-πsirna转染2~4天后,puma的表达显著升高。这些数据表明,含有位于种子区域的脱氧核苷酸组合、2’-氟代脱氧核苷酸、和2’-ome取代核糖核苷酸的gst-πsirna结构出乎意料地使癌细胞的凋亡上升。puma生物标志物的方案如下。在转染前1天,向96孔板中注入细胞(2x103细胞/孔,100μl含有10%fbs的dmem(hyclonecat.#sh30243.01)),在37℃的孵育器(湿润气氛,空气中co2浓度为5%)中培养。第二天,在转染前将培养基置换为90μl含有2%fbs的opti-memi减血清培养基(lifetechnologiescat.#31985-070)。然后,将0.2μl的lipofectaminernaimax(lifetechnologiescat.#13778-100)与4.8μl的opti-memi于室温混合5分钟。将1μl的sirna(储存浓度:1μμ)与4μl的opti-memi混合,加入rnaimax溶液,轻轻混合。将混合物于室温孵育10分钟,形成rna-rnaimax复合物。向每个孔中加入10μl的rna-rnaimax复合物,使sirna最终浓度为10nm。将细胞孵育2小时,并将培养基置换为新鲜的含有2%fbs的opti-memi减血清培养基。在转染后1、2、3、4、6天,用冰冷的pbs洗涤1次细胞,然后用50μl的cell-to-ct裂解液(lifetechnologiescat.#4391851c)于室温裂解5~30分钟。加入5μl的终止溶液,并于室温孵育2分钟。使用taqman通过rt-qpcr测定puma(bbc3,cat#hs00248075,lifetechnologies)mrna水平。实施例8:本发明的gst-πsirna制剂能够使癌异种移植肿瘤在体内显著缩小。以脂质体制剂的形式施予至癌异种移植肿瘤的情况下,gst-πsirna能够在体内发挥基因敲低效能。图7显示了gst-πsirna(序列号61、126)的肿瘤抑制效果。在体内观察到了以gst-π作为靶标的sirna对于gst-πmrna的剂量依赖性敲低。利用的癌异种移植模型使用了以gst-π作为靶标的sirna。gst-πsirna在施予后数天内呈现了显著且出乎意料的有利的肿瘤抑制效果。如图7所示,在以脂质制剂的形式注射4天后,利用gst-πsirna进行的处理导致gst-πmrna表达显著降低。以更高的剂量即4mg/kg注射,24小时后,观测到gst-πmrna表达显著下降约40%。p21sirna以包含组合物(可离子化脂质:胆固醇:dope:dopc:dppe-peg-2k)(25:30:20:20:5)的脂质体制剂的形式单次注射施予,施予量为10ml/kg。对于癌异种移植模型而言,从atcc获得了a549细胞系。在添加有10%胎牛血清、100u/ml盘尼西林和100μg/ml链霉素的rpmi-1640中维持细胞。在接种前48小时稀释细胞,从而使细胞在被收集时处于对数生长期。使用胰蛋白酶-edta轻度消化细胞后,从组织培养物中收集细胞。在台盼蓝的存在下,用血细胞计数器计数并测定活细胞的数量(仅计数活细胞)。在无血清培养基中重新悬浮细胞,使浓度为5x107/ml。然后将细胞悬浮液与冰上融化的bdmatrigel以1:1的比例充分混合,用于注射。小鼠为charlesriverlaboratory的雌性无胸腺裸鼠(nu/nu),经过免疫妥协,6~8周龄,每组3只小鼠。对于肿瘤模型的制备而言,使用25g注射针和针筒,在每只小鼠的右侧腹皮下接种0.1ml含有2.5x106个a549细胞的接种液,每只小鼠接种1剂接种液。小鼠在接种时未被麻醉。对于肿瘤体积的测量和随机化而言,肿瘤大小的测量结果取至0.1mm。肿瘤体积利用下式算出:肿瘤体积=长x宽2/2。每周检测2次肿瘤体积。当培养的肿瘤生长至约350~600mm3时,将小鼠按照各时间点分组。按照给药方案,在同一天施予试验制剂。对于施予量而言,在培养的肿瘤生长至约350~600mm3当天,从4℃冰箱中取出试验制剂。为了形成均质的溶液,在装入注射器之前,手动将装在瓶中的制剂颠倒数次。对于体重而言,小鼠称重至0.1g。每周检测并记录体重2次,包括研究结束当天。对于肿瘤的采集而言,在施予后0、24、48、72、96(可选)、168小时利用高浓度co2杀死动物,并切除肿瘤。先称取肿瘤的湿重,然后将其分成用于分析kd、分配和生物标志物的3部分。用液氮快速冷冻试样,并保存于80℃,直到供于处理为止。本文描述的实施方案不是限制性的,并且本领域技术人员可以容易地理解,可以不进行过度实验而测试本文所述修饰的特定组合以鉴定具有改进的rnai活性的核酸分子。出于所有目的,本文特别提及的所有出版物、专利和文献通过引用整体并入本文。应当理解,本发明不限于所述特定方法、方案、材料和试剂,因为它们可能变化。还应当理解,本文使用的术语仅用于描述特定实施方案的目的,并不意图限制本发明的范围。对于本领域技术人员将显而易见的是,在不背离本说明书的范围和宗旨的情况下,可以对本文所公开的描述进行各种替换和修改,并且这些实施方案在本说明书和所附权利要求的范围内。必须指出,本文和所附权利要求中所使用的单数形式包括复数指称,上下文另有明确说明除外。同样地,术语“一个”(或“一种”)、“一个或更多个(一种或更多种)”和“至少一个/种”在本文可以互换使用。另外要注意,术语“包括”、“包含”、“含有”、“含”和“具有”可以互换使用,并且应该被广义、无限制地解释。除非本文另有说明,否则本文中对数值范围的记载仅仅意在作为对单独提到落入该范围的每个单独的值的速记法,每个单独的值均被并入说明书中,如同它们被单独记载于本文中一样。对于马库什组,本领域技术人员将认识到,该描述包括个体成员以及马库什组成员的亚组。即使没有进一步的阐述,本领域技术人员亦可基于上述描述最大程度地利用本发明。因此,所附的具体实施方案应被解释为仅是说明性的,而不是以任何方式限制本公开的其余部分。本说明书中公开的所有特征可以以任何组合而联用。本说明书中公开的每个特征可以由服务于相同、等同或相似目的的替代特征来代替。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1