一种制备原薯蓣皂苷的方法与流程
本发明属于医药技术领域,涉及化学领域化合物的分离提取方法,具体涉及植物天然药理活性物质原薯蓣皂苷的一种制备方法。
背景技术:
穿山龙是薯蓣科植物穿龙薯蓣Dioscorea nipponica Makino的干燥根茎,性温,味甘、苦,归肝、肾、肺经,具有祛风除湿、舒筋通络、活血止痛、止咳平喘之效。民间多用其泡酒或煎服,治疗风湿弊病、关节肿痛、跌打损伤和闪腰岔气。现代研究表明穿山龙有调节免疫、改善心血管功能、镇咳、平喘、祛痰等多种药理作用,临床用于治疗类风湿性关节炎、冠心病、高血压、心肌硬塞、支气管哮喘。穿山龙中主要有效成分为甾体皂苷,而甾体皂苷主要为薯蓣皂苷和原薯蓣皂苷。
原薯蓣皂苷(protodioscin),分子式为C51H84O22,结构见图1,属于甾体皂苷,又名呋喃固醇皂苷,有很强的药理作用,如增强性功能、降血脂作用、对癌细胞的毒杀作用和抗白血病作用。目前,尚未有一种适合产业化生产同时制备高纯度原薯蓣皂苷的工艺路线披露。市场上原薯蓣皂苷的产量很低,得率较小,难以满足科研或新药研发需求,从而严重限制其深入研究。本发明利用大孔吸附树脂柱层析、反相硅胶柱层析以及反相树脂基柱层析技术,从穿山龙中制备原薯蓣皂苷,纯度好,得率较高,为其开发利用提供良好的物质基础。
技术实现要素:
本发明的目的在于提供一种制备高纯度原薯蓣皂苷的方法,本发明方法不仅操作工艺简单,生产周期短,而且利用本发明可在一个工艺流程中制备得到纯度达到90%以上的原薯蓣皂苷。
为了解决上述问题,本发明提供一种制备原薯蓣皂苷的方法,包括依次以大孔吸附树脂柱层析、反相硅胶柱层析、反相树脂基柱层析的方法来分离穿山龙药材提取液中的原薯蓣皂苷。
一种制备原薯蓣皂苷的方法,包括以下步骤:
1)将干燥的穿山龙药材加入到提取溶剂中,对药材进行提取处理,得到穿山龙提取液,将提取液浓缩、静置、过滤,取滤液;
2)将滤液采用洗脱剂通过大孔吸附树脂柱法进行洗脱处理,收集、合并含有原薯蓣皂苷的洗脱液,得到树脂柱洗脱液;
3)将树脂柱洗脱液浓缩、静置、过滤,得到滤液;
4)将滤液采用洗脱剂通过1-3次反相硅胶柱层析处理,收集、检测、合并洗脱液;
5)将反相硅胶柱洗脱液浓缩,浓缩液采用反相树脂基柱层析处理,收集、检测、合并洗脱液,洗脱液干燥后,得到原薯蓣皂苷。
所述的步骤1)中的提取溶剂选取质量百分比浓度为20%-90%的乙醇溶液,优选为50%-75%含水乙醇溶液。不宜采用水、甲醇或者含水甲醇提取,文献报道原薯蓣皂苷用甲醇回流提取时不稳定,会发生转化反应(参见Kostova I,Dinchev D,Rentsch G H,et al.Z.Naturforsch.57c,33-38(2002))。
所述的步骤1)中的提取处理是通过加热回流来进行提取处理,其中,提取溶剂的体积与药材的重量之比为5-8∶1(即如果药材的重量为1kg,则提取溶剂的体积为5-8L;如果药材的重量为1g,则提取溶剂的体积为5-8mL);提取次数为2-3次,优选为2次;提取时间为2-3h/次;另外,步骤1)中所述的将提取液浓缩是将提取液浓缩至相对密度为1.0-1.2;所述的静置是将浓缩提取液静置过夜(浓缩提取液的相对密度范围以及静置时间有利于杂质沉淀完全,便于后述分离制备)。
所述的步骤2)中的大孔吸附树脂所采用D101、AB-8、DM130H、XAD-2、D201、HP-20、X-5、H103、DM11或ADS-17型树脂中的一种,优选为HP-20、AB-8、D101型大孔吸附树脂;上样比例选择为:大孔吸附树脂的体积与穿山龙药材重量(干重)之比为0.5-1.5∶1,即当穿山龙药材重量为100kg,则大孔树脂柱的柱体积为50-150L,优选为0.7-1.0∶1;进一步的,大孔树脂柱分离处理过程中,大孔树脂柱的柱直径与柱高之比为1∶8-20。
所述的步骤2)中的洗脱剂选依次采用水和含水的乙醇溶液,首先采用水为冲洗液进行冲洗,然后采用质量百分比浓度为20%-50%的乙醇溶液为洗脱剂进行洗脱,收集、合并含有原薯蓣皂苷的树脂柱洗脱液;其中,步骤2)中采用水为冲洗液进行冲洗的过程中,水与大孔吸附树脂的体积之比为2-6∶1,优选为2-3.5∶1;采用所述乙醇溶液为洗脱剂进行洗脱的过程中,乙醇溶液与大孔吸附树脂的体积之比为4-6∶1。
步骤3)中所述将含有原薯蓣皂苷的洗脱液浓缩,将提取液浓缩至相对密度为1.0-1.2;所述的静置,可将浓缩提取液静置过夜(浓缩提取液的相对密度范围以及静置时间有利于杂质沉淀完全,便于后述分离制备)。
所述的步骤4)中的反相硅胶柱层析采用C18反相柱,所述的C18反相柱可选用预装柱或DAC制备柱;其中,步骤4)中所述反相硅胶柱分离,首先采用水为洗脱剂进行洗脱,然后采用质量百分比浓度为10%-50%的乙腈溶液为洗脱剂进行梯度洗脱,收集、合并含有原薯蓣皂苷的洗脱液或者首先采用水为洗脱剂进行洗脱,然后采用质量百分比浓度为20%-60%的乙醇溶液为洗脱剂进行洗脱,收集、合并含有原薯蓣皂苷的洗脱液;其中,步骤4)中第二次反相硅胶柱层析处理过程中,选择乙腈与水的混合溶剂为洗脱剂,所述的乙腈溶液的质量百分比浓度为28%-40%,优选28%-35%。
进行反相柱分离时,采用多次含水乙醇作为洗脱溶剂或含水乙腈作为洗脱溶剂相结合,可进一步提高原薯蓣皂苷的纯度;例如,先使用含水乙醇作为洗脱剂,再使用含水乙腈作为洗脱剂,得到的原薯蓣皂苷纯度可达到90%以上。如再进一步采用反相柱层析,使用含水乙醇或者含水乙腈进行洗脱,可以进一步提高原薯蓣皂苷的纯度。
进一步的,步骤3)和步骤4)中所述的将含有原薯蓣皂苷的洗脱液浓缩,将提取液浓缩至相对密度为1.0-1.2。
反相树脂基填料比传统的C18硅胶具有更高的化学稳定性,可以在强酸、强碱介质下使用,可以用多种有机溶剂作为洗脱剂,可以在较高的浓度下使用,寿命非常长;反相树脂基填料吸附量高,颗粒均匀,机械强度好,不易破碎,残留物少,与相同粒径的C18固定相的柱效相当,无论对极性或非极性样品,几乎无不可逆吸附,非常适用于制备色谱分离纯化微量物质,而C8和C18柱由于硅醇基的存在,样品不可逆吸附到柱子上,使回收率低。
本发明中,对反相硅胶柱层析分离得到的产品进一步采用反相树脂基柱层析,即在所述的步骤5)中的采用反相树脂基柱层析,以进一步提高原薯蓣皂苷的纯度;所选择反相树脂基填料可以为SMB、MCI、GEL、PRP型反相树脂基色谱柱中的一种,柱子规格选择预装柱或或DAC制备柱;其中,步骤5)中所述的反相树脂基柱层析处理过程中选择乙腈与水的混合溶剂为洗脱剂,其乙腈的溶液的质量百分比浓度为15%-35%。
进一步的,所述的穿山龙药材为蓣科植物穿龙薯蓣Dioscorea nipponica Makino的干燥根茎。
由于反相硅胶柱层析使用的反相硅胶粒径对分离效果亦有较大影响,理论上,粒粒径越小的反相硅胶,分离效果越好,但是柱压会明显升高,对设备的要求也相应增加,且反相硅胶的成本也会显著增加,故而本发明优选反相硅胶粒径为40-60μm的反相硅胶。
由于反相树脂基柱层析使用的反相树脂基粒径对分离效果亦有较大影响,理论上,粒径越小的树脂基填料,分离效果越好,但是柱压会明显升高,对设备的要求也相应增加,且填料成本也会显著增加,故而本发明优选粒径为50-70μm的反相树脂基填料。
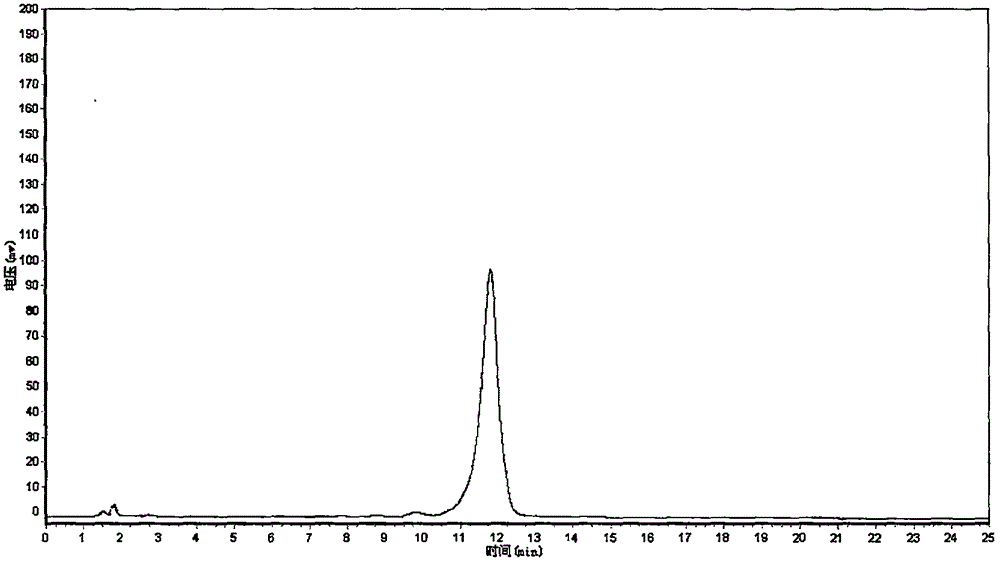
其中原薯蓣皂苷纯度检测如下:样品经高效液相色谱仪在色谱条件为:流动相为乙腈-水(26∶74),波长203nm,色谱柱为Kromasir C18色谱柱(4.6mm×250mm,5μm),流速为1.0mL·min-1,柱温为35℃下检测,样品在HPLC色谱中为单一对称峰,由面积归一化法测定,纯度大于98%,色谱图见图2。
原薯蓣皂苷结构确定的方法如下:使用核磁共振波普仪,对样品进行分析,其中13C-NMR(400MHz,Pyridine-d5)δ:37.5(C-1),30.2(C-2),78.2(C-3),39.0(C-4),140.9(C-5),121.8(C-6),32.4(C-7),31.7(C-8),50.4(C-9),37.1(C-10),21.1(C-11),40.0(C-12),40.8(C-13),56.6(C-14),32.5(C-15),81.1(C-16),63.8(C-17),16.5(C-18),19.4(C-19),40.7(C-20),16.3(C-21),110.7(C-22),37.1(C-23),28.3(C-24),34.3(C-25),75.2(C-26),17.4(C-27);C-3糖分:100.3(GlcC-1′),77.9(GlcC-2′(inner)),78.2(GlcC-3′),79.0(Glc C-4′),76.8(GlcC-5′)61.4(GlcC-6′);102.0(RhaC-1″),72.4(RhaC-2″(1→2)),72.8(Rha C-3″),74.1(RhaC-4″),69.4(RhaC-5″),18.4(RhaC-6″),102.9(Rha C-1″′),72.4(Rha C-2″′(1→2)),72.7(Rha C-3″′),73.9(Rha C-4″′),70.5(Rha C-5″′),18.6(Rha C-6″′);C-26糖部分:104.9(Rha C-1″′),75.2(Rha C-2″′),78.3(RhaC-3″′),71.8(RhaC-4″′),78.6(RhaC-5″′),62.9(RhaC-6″′),13C-NMR和13H-NMR与原薯蓣皂苷的文献报道核磁数据一致,鉴定为原薯蓣皂苷(protodioscin)。
有益效果:本发明的优点包含以下几点:
1)应用本发明方法,可制备得到高纯度的原薯蓣皂苷,原薯蓣皂苷含量达到90-99.9%;
2)应用本发明方法,可得到公斤级的原薯蓣皂苷;
3)本发明制备工艺方法简单,精制效率高,耗能低,环保,操作工艺条件容易控制,质量可控性强;
4)本发明综合反相硅胶柱层析和反相树脂基柱层析的分离技术,得率高,除杂质和色素效果好;
5)本发明所述的高纯度原薯蓣皂苷,无溶剂残留,安全性高,可以单独作为制备药品和保健品的原料,也可与相关药理活性成分相须使用,以达协同增效之功。
附图说明
图1是本发明的原薯蓣皂苷结构图。
图2是本发明的原薯蓣皂苷样品纯度检测色谱图。
具体实施方式
下面通过具体实施例对本发明进行进一步说明:
具体实施例一:
准备干燥的穿山龙药材150kg,粉碎成粗粉后,装入不锈钢中药提取罐中,用质量百分比浓度为70%乙醇溶液,加热回流提取3次,每次8倍量,每次2h;其次,合并提取液,过滤,滤液减压浓缩至相对密度为1.08(40℃),放冷,静置过夜后,过滤,滤液经AB-8大孔吸附树脂柱层析,大孔吸附树脂柱内大孔吸附树脂的柱体积为200L(柱直径0.4m,柱高1.6m),其中柱体积是指大孔吸附树脂装柱后,从柱的底板到树脂沉积表面的体积;树脂柱内树脂的体积与药材重量(干重)之比为4∶3,待滤液完全流入树脂柱后,先用2.5倍柱体积的水洗涤至流出液清澈透明后,再用3倍柱体积的质量百分比浓度为20%的乙醇溶液洗脱洗涤至流出液清澈透明后,然后接着用5倍柱体积的质量百分比浓度为20%的乙醇溶液洗脱,收集洗脱液,洗脱流速为150L/h,每50L洗脱液收集为一流分,液相跟踪检测,合并含原薯蓣皂苷的流份,得到大孔树脂柱洗脱液。将大孔树脂柱洗脱液减压浓缩至相对密度为1.13(40℃),放冷,静置过夜,取上清液;然后将上清液经C18柱(直径500mm,高度800mm,50-70μm)柱层析,分别用水洗脱、25%乙腈洗脱、34%乙腈洗脱,液相跟踪检测,合并含原薯蓣皂苷的流份,浓缩;再进行第二次C18柱(100mm×250mm,50μm)柱层析,分别用25%乙腈和34%乙腈洗脱,液相跟踪检测,合并含原薯蓣皂苷的流份,浓缩;然后利用反相树脂基柱(200mm×250mm,50μm)分离,依次用25%乙腈和30%乙腈洗脱,液相跟踪检测,合并目标流分,减压回收溶剂至干,冷冻干燥,得原薯蓣皂苷0.72公斤(纯度95.2%)。
具体实施例二:
准备干燥的穿山龙药材90kg,粉碎成粗粉后,装入不锈钢中药提取罐中,用质量百分比浓度为50%乙醇溶液,加热回流提取3次,每次8倍量,每次2h。合并提取液,滤过,滤液减压浓缩至相对密度为1.01(40℃),放冷,静置过夜后,过滤,滤液经AB-8大孔吸附树脂柱层析,大孔吸附树脂柱内大孔吸附树脂的柱体积为120L,其中柱体积是指大孔吸附树脂装柱后,从柱的底板到凝胶沉积表面的体积。待滤液完全流入树脂柱后,先用2.5倍柱体积的水洗涤至流出液清澈透明后,再用3倍柱体积的质量百分比浓度为20%的乙醇溶液洗脱洗涤至流出液清澈透明后,然后接着用5倍柱体积的质量百分比浓度为30%的乙醇溶液洗脱,收集洗脱液,洗脱流速为90L/h,每50L洗脱液收集为一流分,液相跟踪检测,合并含原薯蓣皂苷的流份,减压浓缩至相对密度为1.07(40℃),放冷,静置过夜,取上清液;然后将上清液经C18柱(200mm×250mm,50-70μm)柱层析,分别用水洗脱、25%乙腈洗脱、34%乙腈洗脱,液相跟踪检测,合并,合并含原薯蓣皂苷的流份,减压浓缩至相对密度为1.11(40℃),放冷,然后利用反相树脂基柱(200mm×250mm,70μm)分离,依次用20%乙腈和30%乙腈洗脱,液相跟踪检测,合并目标流分,减压回收溶剂至干,冷冻干燥,得原薯蓣皂苷0.4公斤(纯度91.2%)。
具体实施例三:
准备干燥的穿山龙药材120kg,粉碎成粗粉后,装入不锈钢中药提取罐中,用质量百分比浓度为70%乙醇溶液,加热回流提取3次,每次8倍量,每次2h。合并提取液,滤过,滤液减压浓缩至相对密度为1.05(40℃),放冷,静置过夜后,过滤,滤液经AB-8大孔吸附树脂柱层析,大孔吸附树脂柱内大孔吸附树脂的柱体积为160L,其中柱体积是指大孔吸附树脂装柱后,从柱的底板到凝胶沉积表面的体积;待滤液完全流入树脂柱后,先用2.5倍柱体积的水洗涤至流出液清澈透明后,再用3倍柱体积的质量百分比浓度为20%的乙醇溶液洗脱洗涤至流出液清澈透明后,然后接着用5倍柱体积的质量百分比浓度为20%的乙醇溶液洗脱,收集洗脱液,洗脱流速为120L/h,每50L洗脱液收集为一流分,液相跟踪检测,合并含原薯蓣皂苷的流份,减压浓缩至相对密度为1.07(40℃),放冷,静置过夜,取上清液;然后将上清液经C18柱(500mm×800mm,50-70μm)柱层析,分别用水洗脱、35%乙醇洗脱、55%乙醇洗脱,液相跟踪检测,合并含原薯蓣皂苷的流份,浓缩,进行第二次C18柱(500mm×800mm,50-70μm)柱层析,分别用25%乙腈和32%乙腈洗脱,液相跟踪检测,合并含原薯蓣皂苷的流份,减压浓缩至相对密度为1.14(40℃),放冷,然后利用反相树脂基柱(200mm×250mm,70μm)分离,依次用20%乙腈和30%乙腈洗脱,液相跟踪检测,合并目标流分,减压回收溶剂至干,冷冻干燥,得原薯蓣皂苷0.55公斤(纯度96.8%)。
具体实施例四:
首先先通过具体实施例一的步骤得到原薯蓣皂苷0.5公斤,然后用20%乙醇溶解,然后经C18柱(500mm×800mm,50-70μm)柱层析,依次用450L 20%乙醇洗脱、450L 40%乙醇洗脱,750L 50%乙醇洗脱,液相跟踪检测,合并含原薯蓣皂苷的流份,减压回收溶剂至干,冷冻干燥,得原薯蓣皂苷0.43公斤(纯度98.7%)。
以上所述,仅是本发明的较佳实施例而已,并非对本发明的技术范围作出任何限制,故凡是依据本发明的技术实质对以上实施例所作的任何细微修改、等同变化与修饰,均仍属于本发明的技术方案的范围内。
- 还没有人留言评论。精彩留言会获得点赞!