一种淫羊藿次苷I的制备方法与流程
本发明涉及一种淫羊藿次苷I的制备方法,属于食品、保健品、化妆品与医药领域。
背景技术:
淫羊藿次苷I,又名Icariside I,是从中药材淫羊藿中提取分离得到的一种有效单体,其结构如下式(I)所示:
淫羊藿次苷I,是淫羊藿总黄酮中的一种,具有补肾阳,强筋骨,祛风湿的功效。用于肾阳虚衰,阳痿遗精,筋骨痿软,风湿痹痛,麻木拘挛等症状。现代药理学发现此类物质具有抗肿瘤,抗氧化,抗菌消炎,保护心脑血管系统作用。
在CN101891782A的专利文件中公开了淫羊藿次苷I的制备方法。该方法以朝鲜淫羊藿叶为原料,通过乙醇回流提取,粗提物中加入浓盐酸过 滤,滤液中加入蒸馏水析出沉淀,乙醇洗涤沉淀,干燥,得到淫羊藿次苷I。淫羊藿次苷I在不同淫羊藿品种中的含量均很低,一般在0.1%以下,大部分淫羊藿药材中的主要黄酮成分为淫羊藿苷。此方法采用盐酸处理,其目的在于将淫羊藿提取物中的淫羊藿苷通过浓盐酸水解为淫羊藿次苷I。但淫羊藿苷在盐酸作用下,仅有少部分水解为淫羊藿次苷I,绝大部分会彻底水解去掉所有糖基,生成淫羊藿素。另一方面,采用盐酸水解淫羊藿苷,会破坏黄酮环上的异戊烯基,导致水解产物杂质很多。因此,此方法只能通过层析技术才能得到淫羊藿次苷I纯品,使得该工艺不适合工业化生产。
在公开号为CN101899077A的专利文件中公开了淫羊藿次苷I的提取方法及应用。在该方法中,首先用乙醇回流提取淫羊藿叶,再将提取液中加入浓盐酸,加热过滤,再将滤液中加入蒸馏水析出沉淀,因此通过该方法得到的淫羊藿次苷I。然而该方法的缺点也在于采用浓盐酸水解淫羊藿提取液,会破坏黄酮环上的异戊烯基,因此得到的淫羊藿次苷I杂质多,纯度小。
因此需要一种获得高纯度淫羊藿次苷I的方法。
技术实现要素:
本发明的一个目的是提供一种淫羊藿次苷I的制备方法,通过该方法制备得到的淫羊藿次苷I具有转化率高、纯度高和杂质少的优点。
本发明一方面提供了一种淫羊藿次苷I的制备方法,该方法包括以下 步骤:
a.首先将淫羊藿提取物在β-糖苷酶的作用下,进行酶解反应;
b.接着将步骤a得到的酶解反应产物过滤;
c.再将过滤后的滤饼用乙醇结晶,得到所述的淫羊藿次苷I。
优选地,所述步骤a中的淫羊藿提取物中淫羊藿苷质量含量在至少15%。
优选地,在所述的步骤a中,淫羊藿提取物分散于pH值为3.5-7.5的缓冲溶液中,并且酶解反应温度为35-65℃。
优选地,所述的缓冲溶液选自为pH值5.0-6.0的磷酸氢二钠-磷酸二氢钾、乙酸-乙酸钠、磷酸-磷酸钠、柠檬酸-柠檬酸钠和磷酸氢二钠-柠檬酸缓冲溶液中的一种或几种。
最优选地,所述的缓冲溶液体积/L与淫羊藿提取物的质量/Kg比为5-15:1。
优选地,步骤a中所述的酶解反应时间为1-20小时。
更优选地,所述的酶解反应时间为10-20小时。
最优选地,所述的酶解反应时间为15-20小时。
优选地,步骤a中的淫羊藿提取物和β-糖苷酶的重量比为1-50:1。
更优选地,步骤a中淫羊藿提取物和β-糖苷酶的重量比为2-20:1。
最优选地,步骤a中淫羊藿提取物和β-糖苷酶的重量比为5-10:1。
优选地,在所述的步骤c中,首先将过滤后的滤饼干燥,接着进行粉碎,再将粉碎后的滤饼分散于乙醇中回流溶解,过滤,滤液中乙醇的 体积/L相当于粉碎滤饼质量/Kg的10-100倍。
优选地,在所述的步骤c中,将第一次结晶粗品,加入乙醇中回流溶解,过滤,再将滤液冷却再次结晶,回流乙醇的体积/L相当于结晶粗品质量/Kg的20-300倍。
优选地,在所述的步骤a中,所述的淫羊藿提取物通过乙醇溶剂提取淫羊藿中药材制得。
优选地,所述的乙醇溶剂为乙醇水混合溶剂,乙醇水混合溶剂提取淫羊藿中药材得到的提取物首先经过浓缩,接着通过大孔树脂柱吸附,再用体积浓度为30%-100%的乙醇水溶剂进行洗脱,得到用于酶解反应的淫羊藿提取物。
本发明的有益效果在于:1.本发明以淫羊藿提取物为原料,将提取物中的淫羊藿苷选择性的转化为淫羊藿次苷I,转化率达到70%,充分利用了药材资源。2.本发明采用酶解的方法去除淫羊藿苷中的部分糖基,反应条件温和,杂质较少,仅用重结晶的方式便可得到纯度为99%的淫羊藿次苷I,便于工业化大生产。
附图说明
图1上半图表示经过实施例1步骤1.1乙醇提取液中的总黄酮的高效液相色谱图。
图1下半图表示经过实施例1步骤1.2纯化后的乙醇提取液中总黄酮的高效液相色谱图。
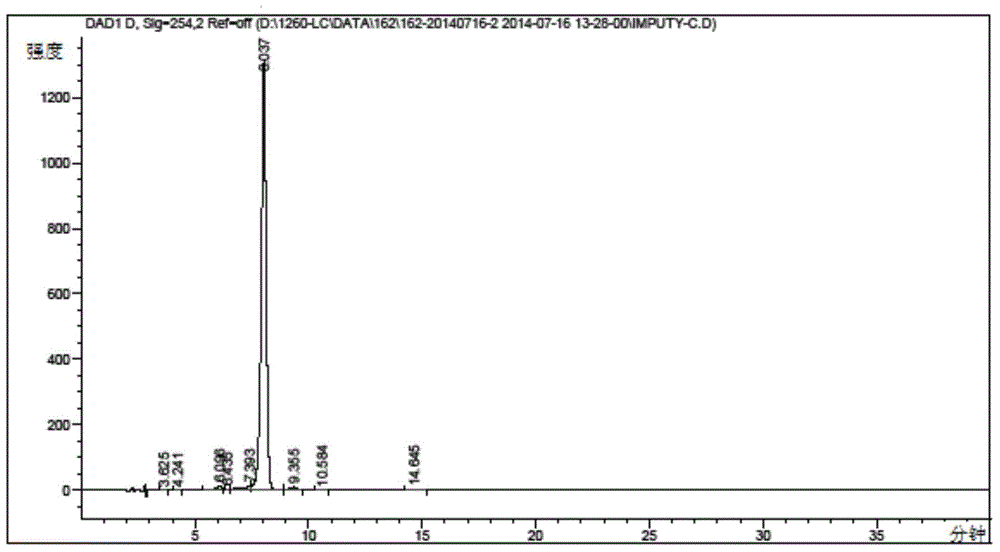
图2表示淫羊藿次苷I纯品的高效液相色谱图。
图3为淫羊藿次苷I纯品的质谱图[M-H]-。
具体实施方式
除非另外说明,本文中的术语“酶解反应”指的是在酶作用下的水解反应。
除非另外说明,本文中术语的“淫羊藿提取物”指的是通过乙醇水溶剂提取淫羊藿得到的提取物,提取物中含有包括淫羊藿苷在内的黄酮类成分以及其他成分。
除非另外说明,本文中的术语“柱床体积”指的是大孔树脂柱内填充物的总体积。
除非另外说明,本文中的术语“β-糖苷酶”为日本天野公司的β-糖苷酶,商品名称为Aromase H2。
实施例1
1.淫羊藿提取物的制备
1.1提取
本实施例分次提取除茎箭叶淫羊藿叶共计400Kg,将其揉碎,分别用14倍、10倍量(v/w)40%体积浓度的乙醇水溶液回流提取2次,每次1小时。回收乙醇至提取液无醇味,通过高效液相色谱检测该提取液中的总黄酮,见附图1上半图。
1.2大孔树脂纯化
将步骤1.1得到的乙醇提取物,浓缩去除乙醇后,用大孔树脂纯化。首先用大孔树脂柱床体积8倍的蒸馏水洗脱大孔树脂柱,蒸馏水洗脱的流速为1倍柱体积/小时,洗脱结束后,弃去洗脱液;
用大孔树脂柱床体积6倍量体积浓度30%乙醇水溶液洗涤大孔树脂柱床,洗脱结束后,弃去洗脱液;
用体积浓度为60%的乙醇水溶剂洗涤步骤2中的大孔树脂柱,乙醇水溶剂体积为柱床体积的4倍,调节乙醇水溶液流速为1倍柱床体积/小时,收集此时的洗脱液。回收乙醇并浓缩至稠膏,喷雾干燥,得到淫羊藿提取物共计9.8Kg。以上三步法得到的大孔树脂纯化后的淫羊藿提取物通过高效液相色谱检测其中的黄酮成分,见图1下半图。
本实施例中的大孔树脂购自河北宝恩吸附材料有限公司,型号为HPD-300型。
高效液相色谱(HPLC)条件:色谱柱:Agela-C18柱;流动相:乙腈-水(其中含体积分数为0.0125%的三氟乙酸),洗脱梯度见下表1,洗脱时间60min,流速:1.0mL/min,柱温:35℃。紫外检测器检测波长分别为254、272、320nm。
表1 溶剂梯度洗脱表
结果显示:除在3分钟时出现的峰为溶剂峰以外,在22分钟、24分钟、46分钟、以及52分钟的出峰均为黄酮成分的峰,通过以上方法检测的药材的总黄酮图谱,与淫羊藿提取物中总黄酮的图谱(HPLC-UV)对比可以看出,经大孔树脂纯化后,淫羊藿提取物中总黄酮成分得到有效富集,其中纯化后的淫羊藿提取物相对于淫羊藿药材,其中总黄酮的质量收率达到2.0~10.0%。
2.酶解反应制备淫羊藿次苷I
称取上述经过大孔树脂纯化的淫羊藿提取物1.0Kg,加入到15L1.0mol/L pH=5.9乙酸-乙酸钠缓冲溶液中,搅拌速度为100rpm情况下使其溶解。待体系温度恒定在60℃条件时,加入0.1Kgβ-糖苷酶搅拌反应。恒温反应3小时后,停止搅拌,反应液降温至30℃。
3.淫羊藿次苷I的纯化
酶解反应结束后,反应液进行离心,收集滤饼,干燥后重量为0.70Kg,粉碎后加入35Kg乙醇,加热回流搅拌1小时后趁热过滤,滤液缓慢降温至25℃后保温10小时,过滤,收集滤饼并干燥,得到淫羊藿次苷I一次结晶产物0.21Kg。将淫羊藿次苷I一次结晶产物粉碎后加入16Kg乙醇,加热回流1小时后过滤,滤液缓慢降温至25℃后保温10小时,过滤,收集滤饼并干燥,得到淫羊藿次苷I重结晶产物共计0.12Kg,其液相色谱和质谱图如图2和图3所示,图2保留时间在6.037min的地方出现的峰为 淫羊藿次苷I的峰,图3出现核质比为529.4的峰,表示[M-H]-,因此通过以上图示表明通过本发明方法获得的淫羊藿次苷I纯度较高。
实施例2
1.酶解反应制备淫羊藿次苷I
称取市购淫羊藿提取物1Kg,加入到15L 0.8mol/L pH=6.0磷酸氢二钠-柠檬酸缓冲溶液中,搅拌速度为100rpm情况下使其溶解。待体系温度恒定在55℃条件时,加入0.2Kgβ-糖苷酶搅拌反应。恒温反应6小时后,停止搅拌,反应液降温至25~30℃。
2.淫羊藿次苷I的纯化
将上述反应液进行离心,收集滤饼,干燥后重量为0.86Kg,粉碎后加入60.2Kg乙醇,加热回流搅拌1小时后趁热过滤,滤液缓慢降温至25℃后保温10小时,过滤,收集滤饼并干燥,得到淫羊藿次苷I一次结晶产物0.26Kg。将淫羊藿次苷I一次结晶产物粉碎后加入20Kg乙醇,加热回流1小时后过滤,滤液缓慢降温至25℃后保温10小时,过滤,收集滤饼并干燥,得到淫羊藿次苷I重结晶产物共计0.15Kg。
- 还没有人留言评论。精彩留言会获得点赞!