一种重复性DNA的合成与组装方法及其应用与流程
本发明涉及一种重复性DNA的合成与组装方法及其应用。
背景技术:
DNA合成与组装是合成生物领域中最重要的支撑技术之一,而特殊性的DNA序列,如高GC、低GC、重复性序列等一直都是工业化DNA合成与组装技术开发中的重点与难点。对于重复性DNA序列的合成与组装,现有最常用的方法是基于酶切连接的常规分子克隆方法与Golden Gate无缝组装方法,以及基于同源重组的Gibson组装方法。
其中,基于酶切连接的常规分子克隆方法在操作可行性上就受到极大的限制,因为重复性DNA序列中极大可能会没有合适的酶切位点,因此,常规分子克隆技术中的酶切连接方法在越来越多的DNA合成与组装应用中被其他方法取代。
鉴于此限制,Golden Gate组装方法得到了开发与应用,因为其消除了上述酶切位点限制,该方法的原理是基于II型限制性内切酶的特征,即切割位点可任意选择,但是识别位点是确定并唯一的,如最最常用的BsaI、BbsI与BsmBI,该方法可不依赖于合成序列中的II性限制性内切酶的存在而使用,在重复性DNA组装过程中有很大优势。但是已经发表的文献中,该方法的操作比较繁琐且构建周期较长,既需要对每个片段进行独立克隆,还需要将每个将供体与载体的抗性进行交叉,这给较长的重复性DNA的合成与组装应用造成了限制;另外,Golden Gate组装方法的供体片段与载体片段都是环状质粒,这给重复性DNA的PCR产物库的合成(因为质粒库有时无法到达PCR产物库的多样性)与重复性DNA合成的PCR产物制备造成了限制,因此,在体外PCR产物库转录等应用中,该方法也有局限性;此外,Golden Gate组装在试剂配置与程序上有多种方法,并且在组装成功率上也有比较大的差异,这也使得其在质粒文库构建中的应用受到了限制。
另外,自2009以来,Gibson组装方法得到了广泛的应用,因为其对酶切位点没有任何依赖性,并且也可以同Golden Gate一样实现多片段的一步组装,因此,在基因合成工业中与合成生物学研究领域得到了深入研究与应用,但是Gibson重组是基于体外重组原理的方法,而重复性DNA片段由于很难设计非常合理的重组臂序列,因此,Gibson重组在该类序列合成中也受到了限制。基于同源重组的Gibson组装方法在进行重复性DNA组装的过程中会发生错误的重组与组装,因此,其在重复性DNA合成与组装过程中也受到极大限制。
与本发明最相似的实现方案是Golden Gate方法,但是本发明优化了该方法所使用的试剂体系与反应程序体系,并且可无缝对接组装产物的PCR扩增体系,使得其在重复性DNA文库的PCR扩增制备与重复性DNA的PCR扩增制备中的应用范围得到了扩展。Golden Gate组装方法的试剂体系不唯一,不同的研究者报道的体系有不同,因此,有时候反应的稳定性得不到保障;本发明优化了试剂体系与反应程序,使得组装反应的稳定性与成功率提高;Golden Gate组装方法的产物需要进行转化筛选等,最终制备形式是质粒或者质粒文库;本发明明确了组装产物的扩增体系与方法,因此拓展了组装产物的最终制备形式,使得其在重复性DNA文库的PCR扩增制备与重复性DNA的PCR扩增制备中得以应用。
技术实现要素:
本发明所要解决的技术问题是克服现有技术的不足,提供一种可靠稳定的重复性DNA的合成与组装方法及其应用。
为解决以上技术问题,本发明采用如下技术方案:
本发明的一个目的是提供一种重复性DNA的合成与组装方法,该方法包括如下步骤:
步骤(1)、重复性DNA序列分析:将需要合成的重复性DNA序列进行酶切位点分析和重复性分析;
步骤(2)、序列拆分与接口选择:根据步骤(1)的分析结果,将所述的重复性DNA序列分为多个亚片段,并确定每个所述的亚片段的接口序列;
步骤(3)、引物的设计:根据步骤(2)的所述的亚片段和所述的接口序列分别对每个所述的亚片段设计并合成引物;
步骤(4)、DNA片段的合成:根据步骤(3)设计并合成的引物进行多个DNA片段的合成;
步骤(5)、重复性DNA的组装:将步骤(4)合成的多个DNA片段、10X T4DNA Ligase Buffer、T4DNA连接酶、II性限制性内切酶以及蒸馏水配置成反应体系,将所述的反应体系进行热循环反应完成重复性DNA产物的组装,其中,所述的热循环的步骤为:
1)、37℃反应4~6分钟;
2)、37℃反应2~4分钟,16℃反应4~6分钟,该步骤循环进行22~28次;
3)、37℃反应8~12分钟;
4)、50℃反应4~6分钟;
5)、80℃反应4~6分钟;
6)、4℃进行保存。
具体地,步骤(1)中,进行重复性DNA序列分析,确保所述的重复性DNA序列内部没有II型限制性酶切位点BsaI、BbsI与BsmBI,同时,找出重复性序列的边界位置,并以此作为步骤(2)中的接口序列选择的依据。
若需要合成的重复性DNA序列不符合步骤(1)的要求,则无法采用本发明进行合成。
具体地,步骤(1)中,采用Lasergene软件套装的genequest模块进行重复性DNA序列分析。
具体地,步骤(2)中,亚片段为2~8个;每个所述的接口序列的内部不能出现镜像重复序列,并且,每两个所述的接口序列没有相同的序列,也没有完全反向互补的序列。
具体地,步骤(3)中,将步骤2中每一段拆分的序列进行接头序列的添加,以确保II型限制性酶切位点的正确性,并在引物末端序列上添加通用的扩增序列。
具体地,步骤(4)中,进行DNA片段合成的方法包括:用长引物作为模板进行通用引物扩增、扩增模板得到、人工基因合成方法获得。
具体地,步骤(5)中,所述的反应体系中各组分的用量为:每个DNA片段各18~22ng;10X T4DNA Ligase Buffer 1.5~2.5μL;T4DNA连接酶0.8~1.2μL;II性限制性内切酶1.3~1.8μL;最后用蒸馏水补足20μL
本发明的另一个目的是提供一种所述的合成与组装方法组装的重复性DNA在重复性DNA文库的PCR扩增制备与重复性DNA的PCR扩增制备中的应用。
本发明的第三个目的是提供一种重复性DNA的PCR扩增制备方法,包括如下步骤:
步骤(1)、根据所述的合成与组装方法组装的重复性DNA产物设计上下游引物;
步骤(2)、将dNTPs混合物、10x Taq Buffer、上下游引物、所述的合成与组装方法组装的重复性DNA产物、Taq酶配置成反应体系,然后将所述的反应体系进行热循环反应完成重复性DNA的PCR扩增,其中,所述的热循环的步骤为:
1)、95℃反应4~6分钟;
2)、95℃反应0.8~1.2分钟,58℃反应0.8~1.2分钟,72℃反应1.5~2.5分钟,该步骤循环进行28~32次;
3)、72℃反应8~12分钟;
4)、4℃进行保存。
优选地,所述的反应体系中各组分的用量为:共计40mM的dNTPs混合物0.8~1.2μL、10x Taq Buffer共4.5~5.5μL、浓度各为10mM的上下游引物各1.8~2.2μL、所述的合成与组装方法组装的重复性DNA产物1.8~2.2μL、Taq酶共1.8~2.2μL。
由于上述技术方案的实施,本发明与现有技术相比具有如下优点:
本发明公开了一种重复性DNA合成与组装的方法及其试剂配方与反应程序,以及其在重复性DNA的PCR产物扩增与重复性DNA的PCR产物文库中的使用方法,较之前的常规限制性内切酶方法、Golden Gate方法与Gibson重组方法等,本发明的方法在重复性DNA合成与组装中更加稳定高效可靠,并且可进行重复性DNA的PCR产物扩增与重复性DNA的PCR产物文库扩增与制备。
Golden Gate组装方法是用一步反应程序,但是本发明使用了热循环程序代替原始一步反应程序,增加了组装反应可靠性与稳定性;Golden Gate组装方法所得产物必须要经过克隆转化,因为无法应用于PCR产物文库的制备过程中,本发明开发了组装产物的扩增体系,扩展了组装产物的适用范围,使得用该方法可制备重复性DNA的PCR产物文库和重复性DNA的PCR产物。
说明书附图
图1为重复性DNA序列分析结果图;
图2为扩增产物测序结果图。
具体实施方式
下面结合具体实施例对本发明做进一步详细的说明,但本发明并不限于以下实施例。本发明中若无特别说明,原料均可市购获得,方法为本领域的常规方法。
文中的“序列”代表着聚合物中单体的排列顺序,比如核苷酸在多核甘酸序列中的排列顺序,此处所说的“核苷酸序列”“核酸”或者“多核苷酸”指的是单链或者双链中的脱氧核糖核苷酸,核苷酸序列是由自然的核苷酸A,T,C,G,U或者其他合成的自然碱基的类似物组成,本发明中,腺嘌呤缩写为A,鸟嘌呤缩写为G,胞嘧啶缩写为C,胸腺嘧啶缩写为T,尿嘧啶缩写为U。多核酸序列可以是单链或者双链,若无特别说明,核苷酸也包括了较长的核苷酸或较短的核苷酸,比如寡核苷酸。
文中采用传统的标记方法来描述多核苷酸序列,单链和左端为5’端,双链的左端为正链5’端。
实施例1:
需要合成与组装的重复性DNA文库序列如下:TRIM文库构建
模板序列如下(SEQ ID NO.1)(划线部分为需要突变的区域,其余部分客户会提供模板RDV,划线部分可以从研发实验室载体PSC03a扩增):
ATCTGCCCCGACACCATCGAAATTAATACGACTCACTATAGGGAGACCACAACGGTTTCCCGAATTGTGAGCGGATAACAATAGAAATAATTTTGTTTAACTTTAAGAAGGAGGTATATCCATGGACTACAAAGACATGCGCGGTAGCCATCATCATCATCATCACGGTAGCGATCTGGGCAAAAAACTGCTGGAAGCAGCTTGGTACGGTCAAGACGACGAAGTTCGCATTCTGATGGCAAACGGCGCAGACGTTAACGCATTTGATACCCGCGGTTGTACGCCGCTCCATCTGGCCGCAAGTTGCGGCCATCTGGAAATCGTCGAAGTCCTGCTGAAAACCGGTGCAGATGTTAACGCTTGGGCTACCGGTCCGATGTTTCAACGCTGCTACGTCACCAACAGCGACTATTTTGGCGAAACGCCGCTGCATCTGGCAGCACCGTTTGGCCATCTGGAAATCGTTGAAGTTCTGCTGAAAGCTGGCGCTGACGTTAATGCACAGGCATTTTCTGGCGTTACCCCGCTGCATCTGGCAGCACGTTGCGGTCATCTGGAAATTGTCGAAGTCCTCCTGAAACACGGCGCAGATGTTAACGCGCAGGATAAAAACGGCTGTACCCCGTTTGATCTGGCAGCAATGTACGGTAACGAAGATATCGCGGAAGTACTGCAGAAAGCGGCAGCTTCCGGAGAATTCCCTCAACCTCCTGTCAATGCTGGCGGCGGCTCTGGTGGTGGTTCTGGTGGCGGCTCTGAGGGTGGCGGCTCTGAGGGTGGCGGTTCTGAGGGTGGCGGCTCTGAGGGTGGCGGTTCCGGTGGCGGCTCCGGTTCCGGTGATTTTGATTATGAAAAGATGGCAAACGCTAATAAGGGGGCTATGACCGAAAATGCCGATGAAAACGCGCTACAGTCTGACGCTAAAGGCAAACTTGATTCTGTCGCTACTGATTACGGTGCTGCTATCGATGGTTTCATTGGTGACGTTTCCGGCCTTGCTAATGGTAATGGTGCTACTGGTGATACCGCACACCTTACTGGTGTGCGG
突变的区域及突变要求
MRGSHHHHHHGSDLGKKLLEAA**GQDDEVRILMANGADVNA*D**G*TPLHLAA**GHLEIVEVLLKTGADVNA#ATG##FQ########YFGETPLHLAA**GHLEIVEVLLKAGADVNA*A**G*TPLHLAA**GHLEIVEVLLKHGADVNAQD**G*TPFDLAA**GNEDIAEVLQKAA
Note:
#:Y—25%;S—25%;G—10%;Others(except Cys)—2.5%;
*:A\D\E\H\K\N\Q\R\S\T----7%;F\I\L\M\V\W\Y---4.3%
E.coli codon optimization used.
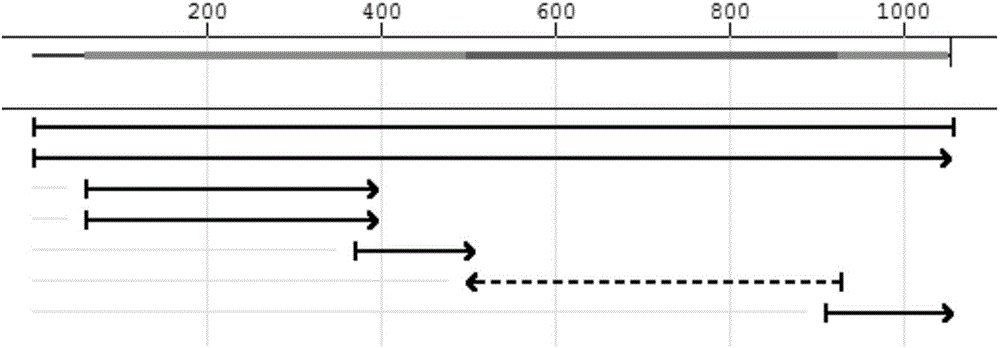
步骤(1)、重复性DNA序列分析:该序列的重复性分析如下,设定重复性序列长度为12bp,结果显示既有正向重复,也有反向重复,序列中没有常用的II限制性内切酶位点BsaI、BbsI与BsmBI,可用本发明的方法进行合成与组装,用Lasergene这个软件套装的genequest模块分析的结果参加图1,其中,最上面的是基因序列长度,一共1100bp左右,第二行I列表示的是Dyad Repeats,是一种短串联连续重复序列;第三行D列表示的是Direct Repeats,是正向重复序列,其中框的长度表示了重复单元的长度;第三列R列表示的Inverted Repeats,是反向重复序列。
步骤(2)、序列拆分与接口选择:该实施例的序列拆分一共分成5段,分别为A、B、C、D、E,其中A与E段有扩增模板,B、C、D是高度可变区序列,并且内部有重复序列;
A(SEQ ID NO.2)(其中斜体部分为接口):
ATCTGCCCCGACACCATCGAAATTAATACGACTCACTATAGGGAGACCACAACGGTTTCCCGAATTGTGAGCGGATAACAATAGAAATAATTTTGTTTAACTTTAAGAAGGAGGTATATCCATGGACTACAAAGACATGCGCGGTAGCCATCATCATCATCATCACGGTAGCGAT
B(SEQ ID NO.3)(其中斜体部分为接口):
CAGCTTGGTACGGTCAAGACGACGAAGTTCGCATTCTGATGGCAAACGGCGCAGACGTTAACGCATTTGATACCCGCGGTTGTACGCCGCTCCATCTGGCCGCAAGTTGCGGCCATCTGGAAATCGTCGAAGTCCTGCTGAAAACCGGTGCAGATGTTAA
C(SEQ ID NO.4)(其中斜体部分为接口):
TGGGCTACCGGTCCGATGTTTCAACGCTGCTACGTCACCAACAGCGACTATTTTGGCGAAACGCCGCTGCATCTGGCAGCACCGTTTGGCCATCTGGAAATCGTTGAAGTTCTGCTGAAAGCTGGCGCTGACGTTA
D(SEQ ID NO.5)(其中斜体部分为接口):
ACAGGCATTTTCTGGCGTTACCCCGCTGCATCTGGCAGCACGTTGCGGTCATCTGGAAATTGTCGAAGTCCTCCTGAAACACGGCGCAGATGTTAACGCGCAGGATAAAAACGGCTGTACCCCGTTTGATCTGGCAGCAATGTAC
E(SEQ ID NO.6)(其中斜体部分为接口):
TACTGCAGAAAGCGGCAGCTTCCGGAGAATTCCCTCAACCTCCTGTCAATGCTGGCGGCGGCTCTGGTGGTGGTTCTGGTGGCGGCTCTGAGGGTGGCGGCTCTGAGGGTGGCGGTTCTGAGGGTGGCGGCTCTGAGGGTGGCGGTTCCGGTGGCGGCTCCGGTTCCGGTGATTTTGATTATGAAAAGATGGCAAACGCTAATAAGGGGGCTATGACCGAAAATGCCGATGAAAACGCGCTACAGTCTGACGCTAAAGGCAAACTTGATTCTGTCGCTACTGATTACGGTGCTGCTATCGATGGTTTCATTGGTGACGTTTCCGGCCTTGCTAATGGTAATGGTGCTACTGGTGATACCGCACACCTTACTGGTGTGCGG。
步骤(3)、接头的添加与引物设计:该实施例的引物设计如下所示:
其中:X1~X32代表的是不同的3个碱基组合,分别如下表1至表3所示:
表1
表2
表3
步骤(4)、DNA片段的制备:该实施例中A/E片段采用可扩增质粒模板,而B/C/D三段均采用重叠引物PCR扩增与长引物作为模板进行PCR扩增得到。
步骤(5)、重复性DNA的组装:将步骤4中获得的片段按照如下体系进行组装反应:每个重复性片段各20ng、10x T4Buffer共2ul、T4DNA连接酶1ul、II性限制性内切酶1.5ul,最后用双蒸水补足至20ul;将该体系进行热循环反应,其中热循环程序如下:37℃反应5分钟、(37℃反应3分钟、16℃反应5分钟,该步骤循环25次)、37℃反应10分钟、50℃反应5分钟、80℃反应5分钟、4℃保存。
步骤(6)、PCR扩增组装产物:按照需要,设计PCR扩增组装产物的上下游引物(如1A-F和1E-R1所示序列),按照以下体系进行扩增反应配置:dNTPs混合物(共40mM)1ul、10x Taq Buffer共5ul、上下游引物各2ul(浓度各为10mM)、步骤5中组装产物2ul、Taq酶共2ul;将该体系进行热循环反应,其中热循环程序如下:95℃反应5分钟、(95℃反应1分钟、58℃反应1分钟、72℃反应2分钟,该步骤循环30次)、72℃反应10分钟、4℃保存。
步骤(7)、扩增产物验证:用电泳方法验证扩增产物大小符合预期,用PCR产物测序结果显示组装反应是正确的,如图2所示,通过测序结果的比对,全长是1100bp左右,下面的测序结果的第4行表示的是全长序列,其下面的所有的都是测序结果比对文件,因为测序结果覆盖到了所有全长区域,所示判定组装正确。
以上对本发明做了详尽的描述,其目的在于让熟悉此领域技术的人士能够了解本发明的内容并加以实施,并不能以此限制本发明的保护范围,且本发明不限于上述的实施例,凡根据本发明的精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!