快速且高效的缀合方法与流程
本申请要求于2015年12月11日提交的美国临时专利申请号62/266,432的优先权,其内容通过整体引用结合以用于所有目的。
背景技术:
对许多基材的聚合物官能化对于它们的应用来说是至关重要的,所述基材包括蛋白质、纳米粒子、固体表面、水凝胶等。例如,由聚合物官能化的蛋白质提供的蛋白质-聚合物缀合物已经广泛用作治疗剂,如PEG基化的(PEGylated)蛋白质。然而,现有的聚合物缀合方法通常制备的官能化的基材具有与反应部位缀合的单一聚合物链。将两种以上不同的个体聚合物链缀合到基材的相同反应部位(以形成所谓的杂臂聚合物、或星形聚合物,其每个具有核心,拥有附接到核心的不同聚合物链的“臂”)是有挑战性的,但对于制造具有多种缀合的聚合物链的组合性能的高度官能化的材料来说却是有力的。例如,两种聚合物链可以缀合到治疗性蛋白的同一活性反应部位,其中一种链延长蛋白的半衰期并且另一种链靶向患病部位。这可以潜在显著地增加药物功效。目前在常规使用中,不存在方便地将多种不同的聚合物链缀合到单一反应部位的方法。本发明致力于这点和其他相关需求。
技术实现要素:
本发明提供了可以用于快速且高效的化学缀合方法的新型方法和组合物,其可以应用于包括聚合物或小分子的改性、连接和缀合在内的各种各样的应用。这种方法还可以用于制造粘合剂材料如水凝胶。
在第一方面中,本发明提供使用儿茶酚官能化的化合物和硫脲官能化的二化合物聚合物(例如,方案1中所示的R1-NCSN-R2)制备多化合物化学结构的方法。所述方法包括这些步骤:(a)在氧化剂的存在下,将所述儿茶酚官能化的化合物(例如,方案1中的R3-Cat)氧化,以形成相应的氧化的儿茶酚官能化的化合物;以及(b)将所述氧化的儿茶酚官能化的化合物与所述硫脲官能化的二化合物聚合物缀合,以形成儿茶酚缀合的三化合物聚合物(例如,方案1中的R1-NCSN-R2-Cat-R3)。在一些实施方案中,所述方法在步骤(b)之后还包括步骤(c):在氧化条件下,将至少一种硫醇官能化的化合物(例如,方案1中的R4-SH或R5-SH)缀合到所述儿茶酚缀合的三化合物聚合物,以形成多化合物化学结构。在一些情况中,在步骤(c)中将两种不同的硫醇官能化的化合物(例如,方案1中的R4-SH和R5-SH)缀合到所述儿茶酚缀合的三化合物聚合物,以形成多化合物化学结构(例如,方案1中的R1-NCSN-R2-Cat-R3-R4和R1-NCSN-R2-Cat-R3-R4-R5)。在一些实施方案中,所述方法还包括在步骤(a)之前的步骤:将儿茶酚分子附接到化合物,以形成所述儿茶酚官能化的化合物。在一些实施方案中,所述方法还包括在步骤(a)之前的步骤:在胺-异硫氰酸酯反应中将具有胺基的化合物与具有异硫氰酸酯基的另一种化合物反应,以形成硫脲官能化的二化合物聚合物。
在一些实施方案中,以上和在本文中描述的方法的步骤(b)在有机溶剂或水性溶剂的存在下进行。在一些实施方案中,以上和在本文中描述的方法的步骤(b)在约1至约14的pH下,更优选在约2至7、或约2至5、或约2.0的pH下进行。在一些实施方案中,氧化剂选自由下列各项组成的组;NaIO4、KIO4、NaMnO4、KMnO4、Na2Cr2O7、K2Cr2O7、CrO3、和蘑菇酪氨酸酶(Mushroom Tyrosinase)。在一些实施方案中,在步骤(b)中,氧化剂与儿茶酚官能化的化合物的比率在0.5至9999之间。
所要求保护的方法的一般示意图在以下方案1中示出,其中R1、R2、R3、R4、和R5独立地彼此相同或不同,R1、R2、R3、R4、和R5中的每一个独立地为小分子、聚合物、大分子(如蛋白质、多核苷酸、脂质等)、纳米粒子、或固体材料的表面。R1、R2、R3、R4、和R5中的任一个均可以是自然存在的或合成性质的化合物。
方案1.制备杂臂聚合物官能化的结构的一般过程
在相关方面中,本发明提供可以在以上和在本文中描述的方法的不同阶段期间存在的各种组合物。第一,本发明提供一种组合物,其包含氧化的儿茶酚官能化的化合物和硫脲官能化的二化合物聚合物(例如,方案1中所示的R1-NCSN-R2)。第二,本发明提供一种组合物,其包含至少一种硫醇官能化的化合物(例如,方案1中的R4-SH或R5-SH)和儿茶酚缀合的三化合物聚合物(例如,方案1中的R1-NCSN-R2-Cat-R3)。在一些情况中,在组合物中存在两种不同的硫醇官能化的化合物(例如,方案1中的R4-SH和R5-SH)。
在第二方面中,本发明提供由以上和在本文中描述的方法制备的多化合物化学结构。多化合物化学结构包含儿茶酚核心和多种共价连结至所述儿茶酚核心的化合物。在一些实施方案中,多化合物化学结构包含共价连结至儿茶酚核心的至少3或4种不同的化合物。在一些情况中,多化合物化学结构具有共价连接至儿茶酚核心的5种不同的化合物(例如,如方案1中示出的R1、R2、R3、R4、和R5)。在一些实施方案中,所述多种化合物中的每一种独立地为小分子、聚合物、大分子、纳米粒子、或固体材料的表面。这些化合物可以是合成的或自然存在的。例如,化合物可以是聚合物如聚酰胺、聚酯、明胶、和透明质酸。
在第三方面中,本发明提供使用儿茶酚官能化的化合物和多臂交联剂制备儿茶酚缀合的聚合物的新型方法,所述方法包括下列步骤:(a)在氧化剂的存在下将所述儿茶酚官能化的化合物氧化,以形成相应的氧化的儿茶酚官能化的化合物;以及(b)将所述氧化的儿茶酚官能化的化合物与所述多臂交联剂缀合,以形成相应的儿茶酚缀合的聚合物。在一些实施方案中,所述氧化剂选自由下列各项组成的组:NaIO4、KIO4、NaMnO4、KMnO4、Na2Cr2O7、K2Cr2O7、CrO3、和蘑菇酪氨酸酶。
在一些实施方案中,制备儿茶酚缀合的聚合物的方法还包括在步骤(a)之前的步骤:将儿茶酚附接到分子,以形成相应的儿茶酚官能化的化合物。在一些实施方案中,制备儿茶酚缀合的聚合物的方法还包括将不同结构的儿茶酚官能化的化合物缀合。
在一些实施方案中,本发明提供制备儿茶酚缀合的聚合物的方法,所述方法还包括在水性或有机溶剂的存在下进行所述步骤(b)缀合。在一些实施方案中,溶剂是水。在一些实施方案中,制备儿茶酚缀合的聚合物的方法还包括在1至14之间的pH下进行所述步骤(b)缀合。在一些实施方案中,步骤(b)在约1至12之间的pH下进行。在一些实施方案中,步骤(b)在约1至8之间的pH下进行。在一些实施方案中,步骤(b)在约2至7的pH下进行。在一些情况中,步骤(b)在约2.0的pH下进行。
在一些实施方案中,制备儿茶酚缀合的聚合物的方法还包括以在0.5至9999、或0.5至5999、或0.5至1999、或0.5至999、或0.5至599、或0.5至199、或0.5至99、或0.5至59、或0.5至19、或0.5至9之间的氧化剂与儿茶酚官能化的化合物的比率进行所述步骤(b)缀合。
在第四方面中,本发明提供上述儿茶酚缀合的聚合物,其包含至少一种多臂交联剂并且连接多种儿茶酚官能化的化合物。在一些实施方案中,所述儿茶酚官能化的化合物还包含附接到分子的儿茶酚。在一些实施方案中,所述儿茶酚官能化的化合物还包含经由连接体附接到分子的儿茶酚。
在一些实施方案中,所述分子是小分子或聚合物。在一些情况中,所述分子是合成的或天然的聚合物。在一些情况中,所述分子选自由下列各项组成的组:聚酰胺、聚酯、明胶、和透明质酸。在一些情况中,所述分子是明胶。在一些实施方案中,所述连接体还包含至少一个硫原子。在一些情况中,连接体还包含至少一个可以用于参与儿茶酚官能化的化合物向多臂交联剂的附接的硫原子。在一些情况中,所述连接体还包含至少一个硫脲基团。在一些情况中,所述儿茶酚官能化的化合物是Gel-NCSN-cat。在一些情况中,所述儿茶酚官能化的化合物是Gel-cat。
在一些实施方案中,多臂交联剂包含具有多个允许与化合物连接的官能化部分的分子。在一些情况中,结构段是聚(乙二醇)(PEG),其中所述PEG的分子量是100至1,000,000。在一些情况中,所述PEG的分子量是1000至1,000,000。在一些情况中,所述PEG的分子量是1000至100,000。在一些情况中,所述PEG的分子量是1000至50,000。在一些情况中,所述PEG的分子量是1000至10,000。在一些情况中,所述PEG的分子量是1000至5,000。在一些情况中,所述多臂交联剂还包含至少一个含有硫原子的官能化部分。在一些情况中,多臂交联剂还包含至少一个可以用于参与儿茶酚官能化的化合物向多臂交联剂的附接的硫原子。在一些情况中,所述多臂交联剂还包含至少一个含有硫脲基团的官能化部分。在一些情况中,多臂交联剂对于每个臂来说包含至少一个硫脲基团。在一些情况中,所述多臂交联剂是硫脲官能化的4臂聚(乙二醇)交联剂(4臂-PEG-NCSN)。在一些情况中,所述多臂交联剂是氨基官能化的4臂聚(乙二醇)交联剂(4臂-PEG-NH2)。
在第五方面中,本发明提供儿茶酚缀合的聚合物形式的示例性的上述杂臂聚合物官能化的结构,如儿茶酚缀合的水凝胶,其能够在被剪切至失效(failure)后恢复粘附性。在一些实施方案中,粘附性的恢复在约0至约14之间的pH下进行。在一些实施方案中,粘附性的恢复在约1至约14之间的pH下进行。在一些实施方案中,粘附性的恢复在约1至约12之间的pH下进行。在一些实施方案中,粘附性的恢复在约1至约8之间的pH下进行。在一些实施方案中,粘附性的恢复在约2至约8,例如约2至约7、或约2至约7之间的pH下进行。在一些情况中,粘附性的恢复在约2.0的pH下进行。如在本文中所使用的,“约”是指参考值的+/-10%。
在第六方面中,本发明提供一种组合物,其包含上述儿茶酚官能化的化合物和氧化剂。
在第七方面中,本发明提供一种组合物,其包含上述氧化的儿茶酚官能化的化合物和一种或多种多臂交联剂。
在第八方面中,本发明提供一种组合物,其包含儿茶酚分子和缀合上述连接体的化合物。
附图说明
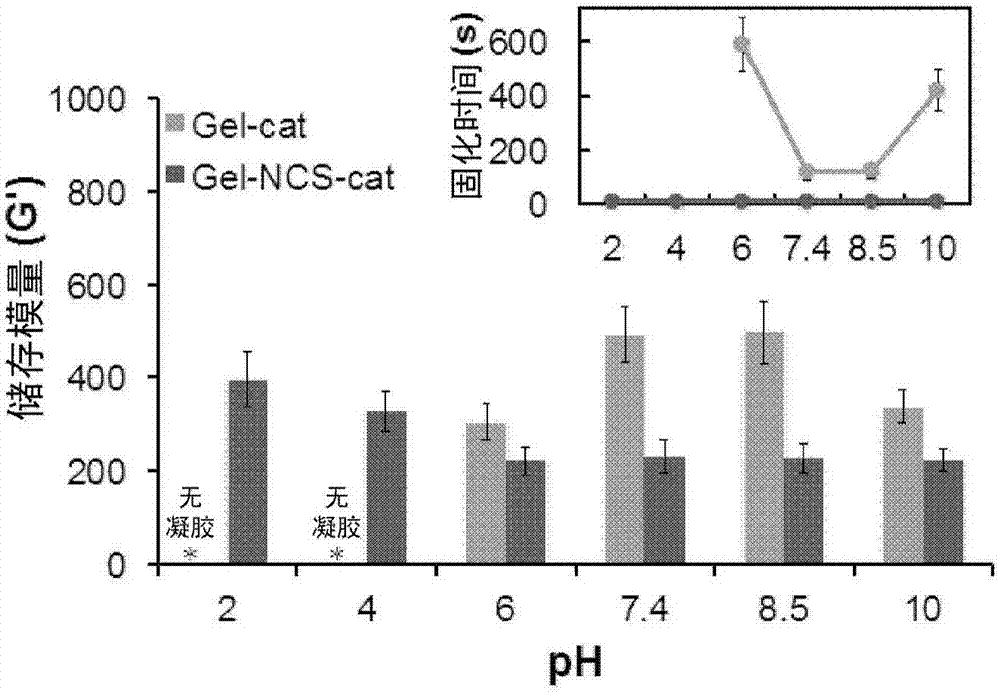
图1A和1B示出了对通过Gel-NCSN-cat和Gel-cat形成的水凝胶的凝胶化行为和机械性能的研究。图1A示出了作为从0.5至6的NaIO4/儿茶酚比率的函数的Gel-cat(红)和Gel-NCSN-cat(蓝)水凝胶的固化时间(插图)和储存模量。(*在NaIO4/儿茶酚低于1的情况下,Gel-NCSN-cat不能形成水凝胶)。图1B示出了作为从2至10的pH的函数的Gel-cat(红)和Gel-NCSN-cat(蓝)水凝胶的固化时间(插图)和储存模量。(*Gel-cat在低pH水平下不能形成水凝胶,并且仅可以在pH=6以上得到流变性数据)。
图2示出了提出的在自然界和生物合成聚合物(利用/不利用人造mfp-6)中的反应途径(a)儿茶酚氧化为醌(b)醌进一步与未氧化的儿茶酚反应以形成二儿茶酚交联剂(c)二儿茶酚进一步与PEG-NH2反应(d)醌迅速与硫脲改性的PEG-NCSN反应。
图3示出了Gel-cat的氧化(NaIO4∶cat=1∶1,pH=6.0)的UV-vis光谱。在加入NaIO4之后立即扫描溶液。箭头表示光谱随时间向前。
图4示出了Gel-cat和PEG-NH2的氧化(NaIO4∶cat=1∶1,pH=6.0)的UV-vis光谱。在加入NaIO4之后立即扫描溶液。箭头表示光谱随时间向前。
图5示出了Gel-cat和PEG-NCSN的氧化(NaIO4∶cat=1∶1,pH=6.0)的UV-vis光谱。在加入NaIO4之后立即扫描溶液。箭头表示光谱随时间向前。
图6A和6B示出了儿茶酚缀合的聚合物的搭接-剪切(lap-shear)粘附强度。图6A示出了通过PEG-NCSN(pH=2.0/7.4)和PEG-NH2(pH=7.4)交联的Gel-cat水凝胶的搭接-剪切粘附强度(n=3)。插图示出了峰粘附应力。图6B示出了通过PEG-NCSN(pH=2.0/7.4)和PEG-NH2(pH=7.4)交联的Gel-cat水凝胶的粘附性恢复。将恢复的粘合强度归一化为初始粘合强度并且以百分比(%)的形式给出(n=3)。
图7示出了在存在和不存在硫脲的情况下以及在不同pH范围下的儿茶酚缀合的聚合物的相对粘附性。
发明详述
I.定义
在本公开中,术语“儿茶酚”当单独使用或以“儿茶酚官能化的”或“儿茶酚缀合的”的形式时,广泛包括苯二酚的全部三种邻位异构体:(1)作为常规命名的化合物的儿茶酚,也被称为焦儿茶酚或1,2-二羟基苯;(2)间苯二酚,或1,3-二羟基苯;和(3)氢醌,或1,2-二羟基苯。化合物可以附接到未被两个羟基占据的苯环的任何碳,从而成为“儿茶酚官能化的化合物”或“儿茶酚缀合的化合物”。例如,R3部分可以连接到在3至6位的碳中的任一个,优选在4或5位。
如在本文中所使用的,术语“儿茶酚缀合的聚合物”是指通过交联剂如多臂交联剂缀合的多种儿茶酚官能化的化合物。儿茶酚官能化的化合物可以是相同或不同的结构。儿茶酚缀合的聚合物的实例包括:水凝胶、有机凝胶以及其他聚合物网络。
如在本文中所使用的,术语“多臂交联剂”是指具有多个允许化合物(包括儿茶酚官能化的化合物)的化学缀合的官能化部分的分子。多臂交联剂还可以具有增加对所附接的官能化部分的空间可达性(accessibility)的结构段或“臂”。例如,多臂交联剂可以是指具有双臂、三臂、四臂或其他多臂结构段附接的官能化部分的分子。此外,结构段可以是具有100至1000000的分子量的聚(乙二醇)(PEG)。例如,多臂交联剂可以是氨基官能化的4臂聚(乙二醇)交联剂(4臂-PEG-NH2)。此外,多臂交联剂还可以具有含有硫原子的官能化部分。多臂交联剂还可以具有包括硫脲基团的官能化部分。多臂交联剂还可以具有全部为硫脲基团的官能化部分。例如,多臂交联剂可以是硫脲官能化的4臂聚(乙二醇)交联剂(4臂-PEG-NCSN)。
如在本文中所使用的术语“氧化剂”和“氧化物”包括具有将其他化合物(特别是儿茶酚)氧化的能力的大范围的化合物。术语氧化物包括但不限于金属氧化物如碱土和过渡金属氧化物。术语氧化物还包括生物氧化剂。示例性的氧化剂包括NaIO4、KIO4、NaMnO4、KMnO4、Na2Cr2O7、K2Cr2O7、CrO3、和蘑菇酪氨酸酶。
如在本文中所使用的,术语“儿茶酚官能化的化合物”是指附接到儿茶酚的分子。分子可以通过连接体附接到儿茶酚。术语分子可以是小分子和包括聚合物在内的大分子。在儿茶酚官能化的化合物的合成中使用的分子包括但不限于:合成的和天然的聚合物,如聚酰胺;聚酯;明胶;凝胶;透明质酸等。连接体还可以含有硫原子。连接体还可以含有硫脲基团。例如,儿茶酚官能化的化合物可以是Gel-NCSN-cat或Gel-cat。
如在本文中所使用的,术语“在剪切失效后恢复粘附性”是指聚合物在被搭接剪切测试至失效后恢复粘附性的能力。可以在通用测试机上以1mm/min的速度在环境条件下测试到达失效的搭接剪切。还可以在酸性pH下在剪切失效之后测量粘附性的恢复。聚合物的粘附性恢复可以在0至100%,例如10至90%、20至80%、25至75%、或30至50%之间的范围内。
II.儿茶酚缀合的聚合物的制备
A.通用
本发明提供了可以用于快速、不依赖pH且高效的化学缀合方法的新方法和组合物,其可以应用于包括大分子或小分子的改性、连接、和缀合在内的各种各样的应用。尤其是,这种方法可以用于制备杂臂聚合物或制造粘合剂材料如水凝胶。例如,本发明提供通过以下方式使用儿茶酚官能化的化合物和多臂交联剂制备儿茶酚缀合的聚合物的新型方法:在氧化剂的存在下将儿茶酚官能化的化合物氧化,以形成相应的氧化的儿茶酚官能化的化合物;以及将氧化的儿茶酚官能化的化合物与多臂交联剂缀合以形成相应的儿茶酚缀合的聚合物。此外,硫原子以及硫脲基团与多臂交联剂和/或儿茶酚官能化的化合物的结合导致在儿茶酚缀合的聚合物中儿茶酚对氧化的增强的稳定性,导致粘附性的明显提高。儿茶酚官能化的化合物可以是相同或不同的结构。
此外,本发明提供用于制备新型水凝胶粘合剂的方法。例如,可以使用硫脲官能化的聚合物模拟mfp-6的功能以用于制备粘合剂水凝胶。不同于通常在碱性条件下反应的天然存在的亲核体如半胱氨酸(pKa=8-9),13、18已知硫脲(pKa=-1)19衍生物由于低pKa值在酸性条件下对儿茶酚衍生物具有反应性。20硫脲基团起出色亲核体的作用以在酸性条件下与儿茶酚缀合的结构形成交联,导致快速形成水凝胶。归因于硫脲的高还原能力,在所得水凝胶粘合剂中儿茶酚针对氧化的稳定性明显增强,因此导致粘附性的明显提高。
B.儿茶酚官能化的化合物的组分和制备
本发明的儿茶酚官能化的化合物包含经由连接体附接到儿茶酚的分子。可以通过经由连接体将儿茶酚分子附接到化合物合成儿茶酚官能化的化合物。
在儿茶酚官能化的化合物的合成中使用的分子包括,但不限于聚酰胺、聚酯、明胶、和透明质酸。在一些实施方案中,选择明胶,一种源自胶原蛋白的多肽的混合物,作为在儿茶酚官能化的化合物的合成中使用的分子。在一些实施方案中,选择水凝胶作为在儿茶酚官能化的化合物的合成中使用的分子。
水凝胶是亲水性的天然或合成聚合物链的网络或支架,有时以其中水是分散介质的胶体状凝胶的形式存在。作为具有高吸水能力的聚合物网络,水凝胶通常严格模拟天然的细胞外基质。归因于较高的水含量,水凝胶还倾向于拥有与天然组织非常相似的柔韧度:在一些情况中,水凝胶可以含有远远超过90%的水。在水凝胶中使用的常用成分包括聚乙烯醇、聚丙烯酸钠、丙烯酸酯聚合物和具有大量亲水基团的共聚物。正在研究天然水凝胶材料以用于组织工程;这些材料包括琼脂糖、甲基纤维素、透明质酸、和其他天然来源的聚合物。示例性的水凝胶可以发现于美国专利号8,329,763;8,008,476;6,534,083;4,438,258;美国专利申请号2013/0,034,592;2013/0,022,569;2002/0,009,591;和国际专利公布号WO/2001/049240;和WO/1997/030092。
可以以多种方式合成儿茶酚官能化的化合物,包括通过常规的碳二亚胺偶联1、21或异硫氰酸酯-胺偶联使含有儿茶酚的分子与含有胺的化合物反应。例如,可以通过在碱性条件下在室温下使胺基(-NH2)与异硫氰酸酯基(-N=C=S)反应得到异硫脲基团(-NH-C(=S)-NH-、或-NCSN-)来合成儿茶酚官能化的化合物(参见方案1)。在氧化条件(高碘酸钠,与儿茶酚基团等摩尔比)下,儿茶酚基团在宽pH范围下迅速与-NCSN-基团反应。
C.多臂交联剂的组分和制备
本发明使用具有多个与官能化部分连接的结构段(或臂)的多臂交联剂。在一些实施方案中,多臂交联剂是指两臂、三臂、四臂或其他多臂衍生物。在一些情况中,结构段可以具有包含一个或多个聚(乙二醇)(PEG)链的支链或直链结构,其中所述PEG的分子量是100至1000000。在一些情况中,多臂交联剂可以具有含有硫原子的官能化部分。在一些情况中,多臂交联剂可以具有硫脲基团。在一些情况中,多臂交联剂对于每个臂来说可以具有硫脲基团。多臂交联剂可以是硫脲官能化的4臂聚(乙二醇)交联剂(4臂-PEG-NCSN)。此外,多臂交联剂可以是氨基官能化的4臂聚(乙二醇)交联剂(4臂-PEG-NH2)。
在本发明中使用的多臂交联剂中的一些,包括两臂、三臂、四臂、六臂、和八臂,是可商购获得的,例如Sigma-Aldrich。此外,可以以多种方式合成在本发明中使用的多臂交联剂,包括通过使可商购获得的多臂交联剂经由异硫氰酸酯-胺偶联进一步功能化以包含硫原子或硫脲基团。例如,使4臂聚(乙二醇)-NH2HCl与异硫氰酸甲酯和三乙胺反应得到相应的硫脲官能化的4臂交联剂。
D.儿茶酚缀合的聚合物的组分和制备
本发明提供制备儿茶酚缀合的聚合物的新型方法。通过以下方式利用多臂交联剂来聚合儿茶酚官能化的化合物:在氧化剂的存在下将所述儿茶酚官能化的化合物氧化,以形成氧化的儿茶酚官能化的化合物,然后,将氧化的儿茶酚官能化的化合物与多臂交联剂缀合,以形成相应的儿茶酚缀合的聚合物。儿茶酚官能化的化合物可以是相同或不同的结构。此外,硫原子以及硫脲基团向多臂交联剂和/或儿茶酚官能化的化合物的合并得到在儿茶酚缀合的聚合物中儿茶酚对氧化的增强的稳定性,导致粘附性的明显提高。
儿茶酚缀合的聚合物可以通过下列方式合成:将儿茶酚官能化的化合物氧化以得到相应的儿茶酚官能化的化合物氧化的醌,接着引入多臂交联剂以得到所需的儿茶酚缀合的聚合物。例如,Gel-cat的氧化得到相应的Gel-醌,其在加入4臂-PEG-NCSN时得到儿茶酚缀合的聚合物Gel-cat+PEG-NCSN。更具体地,可以通过在目标pH值下以1∶1的NaIO4∶NCSN的比率将Gel-cat与4%PEG-NCSN/PEG-NH2交联来形成儿茶酚缀合的水凝胶。
此外,本发明提供杂臂聚合物的制备。杂臂聚合物,在本公开中也被称为“多化合物化学结构”,具有核心结构和与核心连接并且从核心延伸的不同的化合物(例如,聚合物链)。因为对于每个杂臂聚合物来说存在至少两个、可能三条、四条、五条、或更多条链,其中至少一些是彼此不同的(尽管一些其他的可以是相同的),所以这些不同的化合物(在一些情况中为聚合物链)被称为“杂臂”。例如,合成的杂臂聚合物可以通过顺序的单部位偶联反应容易地合成。例如,胺末端官能化的聚合物(聚合物A)与异硫氰酸酯末端官能化的聚合物(聚合物B)之间的偶联得到聚合物缀合物(聚合物A-B),并且异硫脲在偶联部位。然后可以在氧化条件下将儿茶酚末端官能化的聚合物(聚合物C)与异硫脲基团偶联,得到杂臂聚合物(聚合物A-B-C)。硫醇官能化的聚合物(聚合物D和聚合物E)可以在氧化条件下进一步与杂臂聚合物缀合以得到更复杂的结构如聚合物A-B-C-D-E(参见方案2)。
本发明还提供充分限定的利用杂臂聚合物的表面胺的官能化。可以通过杂臂聚合物将具有胺的基材表面官能化。例如,可以将异硫氰酸酯末端官能化的聚合物(聚合物A)偶联到表面胺,得到异硫脲基团,其用于儿茶酚末端官能化的聚合物B的偶联。可以将硫醇官能化的聚合物(聚合物C和聚合物D)在氧化条件下进一步缀合到杂臂聚合物,以在表面上得到更复杂的杂臂聚合物结构如聚合物A-B-C-D(参见方案3)。
本发明还提供充分限定的利用杂臂聚合物的纳米粒子的官能化。可以通过杂臂聚合物将具有胺的粒子表面官能化。例如,可以将异硫氰酸酯末端官能化的聚合物(聚合物A)偶联到表面胺,得到异硫脲基团,其用于儿茶酚末端官能化的聚合物B的偶联。可以将硫醇官能化的聚合物(聚合物C和聚合物D)在氧化条件下进一步缀合到杂臂聚合物,以在纳米粒子表面上得到更复杂的结构如聚合物A-B-C-D(参见方案4)。
本发明还提供不同聚合物向蛋白质的相同的胺的相继缀合。可以通过杂臂聚合物将蛋白质表面上的胺官能化。例如,可以将异硫氰酸酯末端官能化的聚合物(聚合物B)偶联到蛋白质(A)的表面,得到异硫脲基团,其用于儿茶酚末端官能化的聚合物C的偶联,得到蛋白质-聚合物缀合物A-B C。可以将硫醇官能化的聚合物(聚合物D和聚合物E)在氧化条件下进一步缀合到杂臂聚合物,以得到更复杂的结构如蛋白质-聚合物缀合物A-B-C-D-E(参见方案5)。
本发明还提供具有相同交联部位的水凝胶网络的相继交联。例如,胺官能化的4臂聚合物(聚合物A)可以与异硫氰酸酯官能化的4臂聚合物(聚合物B)形成水凝胶,在交联部位留下异硫脲基团。儿茶酚官能化的4臂聚合物(聚合物C)之后可以通过异硫脲基团和儿茶酚基团之间偶联使聚合物网络在相同交联部位进一步交联(参见方案6)。
本发明还提供水凝胶在交联部位的官能化。例如,胺官能化的4臂聚合物(聚合物A)可以与异硫氰酸酯官能化的4臂聚合物(聚合物B)形成水凝胶,在交联部位留下异硫脲基团。儿茶酚官能化的肽(肽C)之后可以通过异硫脲基团和儿茶酚基团之间的偶联在相同交联部位缀合到聚合物网络(参见方案7)。
III.粘附性的测量
为了测量被剪切至失效后粘附性的恢复,可以将儿茶酚缀合的聚合物搭接剪切测试至失效,然后观察并且测量任何粘附性的恢复。儿茶酚缀合的聚合物还可以在酸性pH下在被剪切至失效后恢复粘附性。儿茶酚缀合的聚合物的粘附性恢复可以在0至100%的范围内。儿茶酚缀合的聚合物的粘附性恢复可以高达100%。此外,儿茶酚缀合的聚合物可以多次在被剪切至失效后恢复一些粘附性。儿茶酚缀合的聚合物也可以五次在被剪切至失效后恢复一些粘附性。
实施例
仅作为说明而不是作为限制提供以下实施例。本领域技术人员将会容易地认识到可以被改变或修改以产生基本上相同或相似的结果的多种非关键参数。
实施例1:作为儿茶酚官能化的化合物的Gel-cat聚合物的制备和表征
贻贝足蛋白(Mussel foot proteins,mfp),包括mfp-1、mfp-3和mfp-5,已知是贻贝足中的出色天然粘合剂。L-3,4-二羟基苯丙氨酸(L-DOPA)的儿茶酚侧链是这些使得贻贝能够紧密附接到各种表面的蛋白的关键组分。受这些含有L-DOPA的蛋白质的启发,最近已经合成了许多带有L-DOPA状结构的官能化聚合物,以模拟这样的天然粘合剂。通常,利用DOPA或其儿茶酚类似物(例如,多巴胺)官能化的合成聚合物可以通过氧化被交联从而形成粘合剂水凝胶。1-4已经提出,氧化剂引发的儿茶酚在碱性pH下的聚合是造成水凝胶形成的原因。具体地,儿茶酚首先被氧化为醌。这种中间产物随后引发芳氧基自由基介导的苯酚偶联,2,3引起聚合物交联和水凝胶形成。然而,由于儿茶酚结构的丧失,这样的醌介导的交联不可避免降低粘附性。5,6之前的研究已经显示,在碱性条件下的醌形成降低了儿茶酚官能化的水凝胶与云母、7-9二氧化钛10和二氧化钛氧化物表面的粘附性。6、11、12因此,为了更好地保持粘附性,应当保护儿茶酚结构免于氧化,并且这在本质上与模拟mfp的聚合物的交联条件是矛盾的。1-3
在自然界中,贻贝通过采用富含硫醇的mfp-6作为具有非常低的pKa(pKa=3-4)的还原剂来克服儿茶酚氧化的负面影响13、14。mfp-6的硫醇盐可以在酸性pH(pH≤5)下7、13、16将醌6、13-15还原为原始儿茶酚结构,因此恢复甚至提高向基材的粘附性。13、16、17除了作为天然抗氧化剂,13、14、17值得注意的是mfp-6对于提高斑块蛋白之间的内聚力来说也是关键的。13、15鉴于此,可以在聚合物水凝胶粘合剂的结构设计中采用“人造mfp-6”。“人造mfp-6”应当拥有三个关键性质:(i)具有低pKa,从而可以在酸性pH(pH≤5)下进行交联,(ii)能够将醌还原为儿茶酚(为了更好的粘附性),和(iii)含有强亲核体(如硫醇),从而其可以有效地与醌相互作用以使水凝胶的聚合物网络交联(为了更好的内聚力)。
选择明胶,一种源自胶原蛋白的多肽的混合物,作为聚合物骨架。为了模拟富含DOPA的mfp(mfp-1、mfp-3和mfp-5),通过常规的碳二亚胺偶联1、21来合成儿茶酚官能化的明胶。碳二亚胺偶联得到酰胺连结的儿茶酚官能化的明胶聚合物,被称为Gel-cat,(1,图1A)。21与之前的报道一致,可以通过仅在碱性条件下氧化使Gel-cat交联。通过1H NMR和UV-vis测量估算明胶的儿茶酚含量。为了对照两种不同聚合物中存在的儿茶酚基团的量,在整个研究中,除非另外指定,使用10%Gel-cat制备对照水凝胶,而使用4%的Gel-NCSN-cat制备用于比较的水凝胶。通过儿茶酚在不同氧化剂含量下交联,形成水凝胶。首先,将Gel-cat溶解于PBS中,Gel-cat的浓度是100mg/mL,从而设定等摩尔量的儿茶酚。然后,将NaIO4加入并且保持在相对于儿茶酚0.5-6的摩尔比/pH水平为pH=6至pH=10。将混合物立即涡旋并且当在将瓶反转之后聚合物溶液停止流动时测定凝胶化时间。
实施例2:作为儿茶酚官能化的化合物的4Gel-NCSN-cat聚合物的制备和表征
为了模拟富含DOPA的mfp(mfp-1、mfp-3和mfp-5),通过异硫氰酸酯-胺偶联来合成儿茶酚官能化的明胶。首先合成多巴胺-异硫氰酸酯22、23,然后将其经由异硫氰酸酯-胺偶联接枝到明胶化合物,从而制备儿茶酚官能化的化合物Gel-NCSN-cat(2,图1A)。Gel-NCSN-cat的硫脲连接体模拟了mfp-6的还原性硫醇。通过1H NMR和UV-vis测量估算明胶的儿茶酚含量。通过儿茶酚在不同氧化剂含量下的交联,形成水凝胶。首先,将Gel-NCSN-cat和Gel-cat溶解于PBS,Gel-NCSN-cat和Gel-cat的浓度分别是40mg/mL和100mg/mL,从而设定等摩尔量的儿茶酚。然后,将NaIO4加入并且保持在相对于儿茶酚1-6的摩尔比/pH水平为pH=2至pH=10。将混合物立即涡旋并且当在将瓶反转之后聚合物溶液停止流动时测定凝胶化时间。
实施例3:4臂PEG-NCSN-CH3的制备和表征
将4臂聚(乙二醇)-NH2HCl盐(sat)溶解于干燥DCM中。在溶液中加入异硫氰酸甲酯和三乙胺。将混合物倒入冷二乙基醚中并且通过离心机和真空干燥得到PEG产物。通过在1H NMR光谱中相关峰的积分测定异硫氰酸甲酯的接枝比率。
实施例4:在不同的NaIO4:儿茶酚比率下的水凝胶(Gel-NCSN-cat和Gel-cat)的制备
通过儿茶酚在不同氧化剂含量下交联,形成水凝胶。首先,将Gel-NCSN-cat和Gel-cat溶解于PBS中。然后,将NaIO4加入并且保持在相对于儿茶酚0.5-6的摩尔比。将混合物立即涡旋并且当在将瓶反转之后聚合物溶液停止流动时测定凝胶化时间。
实施例5:在不同的pH值下的水凝胶(Gel-NCSN-cat和Gel-cat)的制备
通过在不同的pH条件下使交联儿茶酚来形成水凝胶。首先,将Gel-NCSN-cat和Gel-cat溶解于pH缓冲剂中(2,HCl;4,MES;6,PBS;7.4,PBS;8.5,PBS;10,NaOH)。然后以相对于儿茶酚1.0的摩尔比加入NaIO4。由于它们的短固化时间而将混合物立即涡旋并且当在将瓶反转之后聚合物溶液停止流动时测定凝胶化时间。
实施例6:儿茶酚缀合的聚合物的制备和表征
为了研究硫脲连接对Gel-NCSN-cat凝胶化的贡献,合成了硫脲官能化的4臂聚(乙二醇)交联剂(PEG-NCSN),其在氧化条件下使Gel-cat交联。如预期的,在所有测试的pH值下,凝胶化都是快速的(在1min内)。然而,加入了氨基官能化的4臂聚(乙二醇)(PEG-NH2)的阴性对照与Gel-cat仅在较高pH水平(pH≥6)下以明显更长的固化时间形成水凝胶。这样的差异可以归因于硫脲与氨基相比较低的pKa,并且还表明硫脲是高度有效的用于与氧化的儿茶酚(醌)偶联的亲核体。
Gel-cat的凝胶化时间表明了对氧化剂含量(NaIO4和儿茶酚之间的摩尔比)和pH值二者的强的依赖性(图1A和1B中的红色曲线)。这些观察结果与之前的报道2、3、24一致,即氧化剂含量和pH二者是造成儿茶酚的还原(儿茶酚)和氧化(醌)形式之间的化学计量比的原因。值得注意的是,针对在这样的情况中的快速凝胶化,需要儿茶酚和醌之间的最佳比率。鲜明的对比是,Gel-NCSN-cat的凝胶化时间未显示出对氧化剂含量或pH值的明显的依赖性。无论氧化剂含量和pH是多少,对于Gel-NCSN-cat来说,均可以实现极其快速的凝胶化(固化时间≤3s)(参见图1A和1B中的蓝色曲线)。此外,动态流变研究显示,所得水凝胶的机械性能显示出相似的对凝胶化条件的依赖性。具体地,Gel-cat水凝胶的储存模量显示出随着逐渐增加的氧化剂含量或pH值的最初增加和随后的略微降低(图1A和1B中的红色柱),支持了如下的想法:最佳的氧化剂含量和pH值可以得到由被儿茶酚官能化的聚合物组成的水凝胶网络的经由酰胺连接的更快和更致密的交联。2、3相比之下,Gel-NCSN-cat水凝胶的储存模量显示出可忽略的对氧化剂含量和pH值的依赖性(图1A和1B中的蓝色柱)。有趣的是,即使在妨碍Gel-cat的凝胶化的酸性条件(pH=2、pH=4)下,Gel-NCSN-cat也可以形成强得多的水凝胶。这种所得水凝胶的凝胶化行为和机械性能的明显差异可能表明两种儿茶酚缀合的聚合物的两种不同的交联机制。考虑到相同的明胶主链,Gel-cat和Gel-NCSN-cat水凝胶之间的这样的明显差异很可能归因于明胶主链和儿茶酚基团之间的不同连接。
实施例7:儿茶酚缀合的聚合物的机制
为了更好地理解凝胶化机制,使用UV-vis光谱法监测醌的产生和消耗。首先,评价Gel-cat在略微酸性条件(pH=6)下的UV光谱(图3),因为醌在碱性pH25下是高度反应性的且不稳定的。在加入NaIO4(NaIO4∶cat=1∶1)时,醌峰(在395nm2,3)立即出现并且在之后的30分钟内略微升高。同时,可以观察到来源于二儿茶酚形成2,3,26的在281nm和484nm附近的两个新的峰((a)和(b),图2)。在pH 7.4下,由于在该pH下的更快的反应,二儿茶酚峰更明显。如预期的,PEG-NH2(作为阴性对照)的存在将不会改变醌和二儿茶酚形成的过程((c),图2)。相比之下,PEG-NCSN的存在迅速消耗醌并且大幅抑制二儿茶酚结构的形成。如在图4中所示,在加入NaIO4时,醌峰立即出现并且然后在之后的10min内降低。在多至45min内几乎不能观察到二儿茶酚峰。同时,在287nm附近的新的峰出现,据信这是硫脲取代的儿茶酚的吸收。27、28在更酸性的条件(pH 4.0或2.0)下,醌被还原性硫脲的快速消耗变得更明显。快速的醌消耗表示如在小分子之间的反应中所示的将醌恢复为儿茶酚的有效的硫脲-醌偶联20。这样的有效偶联可能归因于硫脲基团的低pKa和出色的亲核性,导致快速形成水凝胶并且保留大量的儿茶酚。
Gel-NCSN-cat的这种凝胶化机制与其中mfp-6在减少儿茶酚的氧化和防止贻贝足的粘合性能的丧失方面起到重要作用的天然贻贝蛋白的凝胶化机制相似。13-15、17、29因此,据信,硫脲官能化的聚合物可以模拟mfp-6,以用于儿茶酚的恢复和出色的水凝胶粘合剂的形成((d),图2)。
实施例8:搭接-剪切测量
为了检验水凝胶的粘附性,进行两个搭接-剪切测量。针对水凝胶制备,我们使用Gel-cat(10%)作为人造的富含DOPA的mfp(mfp-1,3,5)和PEG-NCSN(4%)作为人造的mfp-6(不具有DOPA)。此外,我们研究了Gel-cat和PEG-NH2(4%)的组合作为阴性对照。在通用测试机上以1mm/min的速度在环境条件下对所有样品进行搭接剪切测试至失效。进行至少五次相继的粘合强度测量以研究水凝胶的粘附性能的恢复。4在下一次测试之前,将所有测试水凝胶浸入DI水中3。然后以相同方式测量搭接-剪切强度。尤其是,可以在酸性pH下由PEG-NCSN(人造mfp-6)交联的Gel-cat水凝胶实现最高的粘附强度(pH=2.0,~21kPa,图6A)。在pH=7.4下,搭接-剪切强度降低至仅2kPa。这可能是由于硫脲在碱性条件下受损害的还原能力导致的。然而,由PEG-NCSN交联的水凝胶的粘合强度仍然高于PEG-NH2交联的水凝胶的粘合强度(~1KPa),表明在pH 7.4下硫脲介导的交联机制仍然超过醌介导的机制。
实施例9:粘附性的恢复
PEG-NCSN(人造mfp-6)交联的Gel-cat水凝胶可以在酸性pH下在被剪切至失效后部分地恢复粘附性。明显地,第一次恢复接近100%(图6B),并且在5次搭接-剪切测量之后,PEG-NCSN水凝胶仍然可以恢复初始粘附强度的40%。然而,这样的粘附性的自发恢复随着逐渐增加的pH(pH 7.4)而降低。相比之下,无论pH是多少,PEG-NH2交联的水凝胶在第一次搭接-剪切测量之后均未显示出粘附性能的恢复。这样的差异表明,硫脲系水凝胶的粘附性由保留的儿茶酚和表面之间的可逆氢键合介导。
总而言之,描述了提高儿茶酚缀合的明胶水凝胶的粘附性能的新策略。第一次采用了用于聚合物交联和水凝胶制备的硫脲-醌偶联。硫脲在酸性pH下的这样的有效偶联反应和还原能力明显提高了水凝胶粘附性。本发明还可以应用于DOPA研究(如表面涂布)和聚合物化学(如点击化学和蛋白质缀合)的许多其他领域。
本申请中引用的所有专利、专利申请、和其他出版物(包括GenBank登录号)通过引用整体结合在本文中用于所有目的。
参考文献
(1)Hong,S.;Yang,K.;Kang,B.;Lee,C.;Song,I.T.;Byun,E.;Park,K.I.;Cho,S.W.;Lee,H.Advanced Functional Materials 2013,23,1774.
(2)Cencer,M.;Liu,Y.;Winter,A.;Murley,M.;Meng,H.;Lee,B.P.Biomacromolecules 2014,15,2861.
(3)Lee,B.P.;Dalsin,J.L.;Messersmith,P.B.Biomacromol-ecules 2002,3,1038.
(4)Kim,B.J.;Oh,D.X.;Kim,S.;Seo,J.H.;Hwang,D.S.;Masic,A.;Han,D.K.;Cha,H.J.Biomacromolecules 2014,15,1579.
(5)Danner,E.W.;Kan,Y.;Hammer,M.U.;Israelachvili,J.N.;Waite,J.H.Biochemistry 2012,51,6511.
(6)Anderson,T.H.;Yu,J.;Estrada,A.;Hammer,M.U.;Waite,J.H.;Israelachvili,J.N.Advanced functional materials 2010,20,4196.
(7)Kan,Y.;Danner,E.W.;Israelachvili,J.N.;Chen,Y.;Waite,J.H.2014.
(8)Wei,W.;Yu,J.;Broomell,C.;Israelachvili,J.N.;Waite,J.H.Journal of the American Chemical Society 2012,135,377.
(9)Yu,J.;Wei,W.;Danner,E.;Israelachvili,J.N.;Waite,J.H.Advanced Materials 2011,23,2362.
(10)Lee,H.;Scherer,N.F.;Messersmith,P.B.Proceedings of the National Academy of Sciences 2006,103,12999.
(11)Hwang,D.S.;Harrington,M.J.;Lu,Q.;Masic,A.;Zeng,H.;Waite,J.H.Journal of materials chemistry 2012,22,15530.
(12)Yu,J.;Wei,W.;Menyo,M.S.;Masic,A.;Waite,J.H.;Israelachvili,J.N.Biomacromolecules 2013,14,1072.
(13)Yu,J.;Wei,W.;Danner,E.;Ashley,R.K.;Israelachvili,J.N.;Waite,J.H.Nature chemical biology 2011,7,588.
(14)Nicklisch,S.C.T.;Waite,J.H.Biofouling 2012,28,865.
(15)Zhao,H.;Waite,J.H.Journal of Biological Chemistry 2006,281,26150.
(16)Martinez Rodriguez,N.R.;Das,S.;Kaufman,Y.;Israela-chvili,J.N.;Waite,J.H.Biofouling 2015,31,221.
(17)Nicklisch,S.C.T.;Das,S.;Martinez Rodriguez,N.R.;Waite,J.H.;Israelachvili,J.N.Biotechnology progress 2013,29,1587.
(18)Harris,T.K.;Turner,G.J.IUBMB life 2002,53,85.
(19)Schiessl,W.C.;Summa,N.K.;Weber,C.F.;Gubo,S.;Dücker-Benfer,C.;Puchta,R.;van Eikema Hommes,N.J.R.;van Eldik,R.Zeitschrift für anorganische und allgemeine Chemie 2005,631,2812.
(20)Abdel-Mohsen,H.T.;Conrad,J.r.;Beifuss,U.The Jour-nal of organic chemistry 2013,78,7986.
(21)Wang,X.;Li,Z.;Shi,J.;Wu,H.;Jiang,Z.;Zhang,W.;Song,X.;Ai,Q.ACS Catalysis 2014,4,962.
(22)Medintz,I.L.;Stewart,M.H.;Trammell,S.A.;Susumu,K.;Delehanty,J.B.;Mei,B.C.;Melinger,J.S.;Blanco-Canosa,J.B.;Dawson,P.E.;Mattoussi,H.Nature materials 2010,9,676.
(23)Ji,X.;Palui,G.;Avellini,T.;Na,H.B.;Yi,C.;Knappen-berger Jr,K.L.;Mattoussi,H.Journal of the American Chemical Society 2012,134,6006.
(24)Liu,Y.;Meng,H.;Konst,S.;Sarmiento,R.;Rajachar,R.;Lee,B.P.ACS applied materials&interfaces 2014,6,16982.
(25)Xu,H.;Nishida,J.;Ma,W.;Wu,H.;Kobayashi,M.;Otsu-ka,H.;Takahara,A.ACS Macro Letters 2012,1,457.
(26)Graham,D.G.;Jeffs,P.W.Journal of Biological Chemis-try 1977,252,5729.
(27)Land,E.J.;Perona,A.;Ramsden,C.A.;Riley,P.A.Or-ganic&biomolecular chemistry 2009,7,944.
(28)Borovansky,J.;Edge,R.;Land,E.J.;Navaratnam,S.;Pavel,S.;Ramsden,C.A.;Riley,P.A.;Smit,N.P.M.Pigment cell research 2006,19,170.
(29)Zhao,H.;Waite,J.H.Biochemistry 2005,44,15915.
- 还没有人留言评论。精彩留言会获得点赞!