一种比卡鲁胺中间体的制备方法与流程
本发明属于生物医药领域的化学药物合成,具体涉及一种抗雄激素类药物比卡鲁胺中间体的制备方法。
背景技术:
比卡鲁胺(Bicalutamide),化学名为N-[4-氰基-3-(三氟甲基)苯基]-3-(4-氟苯硫酰基)-2-甲基-2-羟基丙酰胺,英文名为N-[4-cyano-3-(trifluoromethyl)phenyl]-3-[(4-flurophenyl) sulfonyl]-2-hydroxy-2-methyl-propanamide,化学式如下所示:
1995年在英国率先上市,商品名为Casodex,我国于1999年批准进口比卡鲁胺片剂。比卡鲁胺为美国药典32版收载的疗效确切,作用特异性强,口服有效,给药方便,耐受性好,且有较长的半衰期的一种非甾体类抗雄激素药物,是目前唯一口服一次(50mg/片)治疗前列腺癌的药物,与LHRH 合用可应用于晚期前列腺癌的治疗,与第一代抗雄激素药物氟他胺相比可减少各种毒副作用70%,并且效果要高于氟他胺,是目前销售额增长最快的抗肿瘤药物,具有良好的临床应用前景,国内自主研制比卡鲁胺,可以降低药品价格,减少外汇,满足人民用药需要。
文献报道的比卡鲁胺的合成方法很多,有实用价值的路线是由4-氨基-3-三氟甲基苯胺与2-甲基丙烯酰氯缩合成酰胺,再经氧化得环氧化物,开环与4-氟苯硫酚得到比卡鲁胺重要的硫酚中间体,进而氧化得比卡鲁胺(Howard Tucker J.Med.Chem.,1988, 31 ,954-959;Howard Tucker,USP4636505),合成路线如下:
左旋比卡鲁胺的生理活性要比右旋的异构体强60 倍,因此不对称合成左旋异构体尤为重要。左旋比卡鲁胺的合成路线(Leonid Kirkovsky.et al. J.Med.Chem., 2000,43,581-590)是由D-脯氨酸与2-甲基丙烯酰氯缩合成酰胺,NBS溴化,酸化开环后与4-氨基-3-三氟甲基苯胺得酰胺,再与4-氟苯硫酚生成 (R)-硫酚中间体,最后经氧化得到左旋比卡鲁胺,合成路线如下:
另外还有许多专利(WO0128990,WO134563, WO9519770和US5985868)也有相似制备左旋比卡鲁胺的报导。
从上述主要两条路线中看以看出,制备比卡硫醚两个重要的中间体是环氧化合物(Ⅰ)和卤代醇化合物(Ⅱ),化学式如下所示:
(Ⅰ)
(Ⅱ)
其中环氧化合物(Ⅰ)制备的报道很多,有O. Payen et al.;J. Organomet. Chem., 2011,696,1049-1056; K.M. Schragl et al., Tetrahedron Letters , 2013,54,2239–2242; Q. Shi et al., Bioorg. Med. Chem., 2012,20,4020–4031; Howard Tucker J.Med.Chem.,1988, 31 ,954-959等;
卤代醇化合物(Ⅱ)主要是通过下面两条路线进行合成(WO2010116342,US2005085449,WO2009036206,WO2007027582,WO2009155481,WO2013067170等):
这两条路线中,第一条路线较长,周期长,收率中等,并使用了甲磺酰氯管制试剂和烯烃双羟基化试剂(大多为重金属试剂,如四氧化锇,高锰酸钾等),造成极大的环境污染,不利于工业化生产;第二条的原料是一个油状物较难获得,合成路线较长,且提纯难。
为了尽可能克服以上路线中存在的问题,本发明人设计出合成卤代醇化合物(Ⅱ)的新型工艺路线,并通过实验证明此路线的可行性。此路线具有起始物料易得,反应收率高、反应易操作等优点,具有广泛的工业应用前景。
技术实现要素:
本发明的技术方案和内容涉及一种比卡鲁胺中间体的制备方法,该比卡鲁胺中间体如下式(Ⅲ)所示:
(Ⅲ)
其目的在于:提供一条可适用于大规模生产的制备比卡硫醚中间体的新方法,从而解决比卡鲁胺现有合成技术中存在的安全性差,污染环境,成本高等问题。
一种比卡鲁胺中间体的制备方法,具体地讲是一种用N-(4-氰基-3-三氟甲基苯基)甲基丙烯酰胺制备3-氯-N-(4-氰基-3-三氟甲基苯基)-2-羟基-2-甲基丙酰胺的方法,其合成路线图如下:
具体步骤如下:
在溶剂中,以N-(4-氰基-3-三氟甲基苯基)甲基丙烯酰胺为原料,加入酸,冷却到0~15℃,搅拌,滴加入次氯酸钠溶液氧化,滴毕保持反应体系温度在0~15℃,反应进行0.5~1小时,得到含有粗品的溶液,经分离纯化,得到高纯度的化合物(Ⅲ);
其中所述溶剂可选用任何与水互溶的一种或几种溶剂的混合物,如水、丙酮、甲醇、乙醇、DMSO 、DMF 、DMA、二氧六环、乙腈等,优选为丙酮;所述溶剂与N-(4-氰基-3-三氟甲基苯基)甲基丙烯酰胺的体积重量比为10 ~ 20:1,优选为15:1;
其中所述酸可任选为盐酸,硫酸,冰醋酸或磷酸中的一种,优选为硫酸;所述酸与N-(4-氰基-3-三氟甲基苯基)甲基丙烯酰胺的摩尔用量比为1 ~ 3:1,优选为1.5:1;
其中所用氧化剂为市面上常用的次氯酸钠溶液(漂白剂),其质量百分比浓度为10~13%,优选质量百分浓度为10%;次氯酸钠与N-(4-氰基-3-三氟甲基苯基)甲基丙烯酰胺的摩尔用量比为1 ~ 5:1,优选为2:1;
其中所述冷却温度和反应体系温度为0~15℃,优选0~5℃;
其中所述分离纯化是将得到含有粗品的溶液,经萃取,分液,洗涤,无水硫酸钠干燥,过滤,甲苯重结晶;所述萃取用的萃取液包括乙酸乙酯、二氯甲烷、乙酸异丙酯或乙酸丁酯,优选为乙酸乙酯。
本发明有反应易于操作,条件温和,后处理简便,无环境污染,成本低等优点,且收率基本可达到75-90%,纯度可达到97%以上。最突出的优点:一是反应时间大大缩短,用N-(4-氰基-3-三氟甲基苯基)甲基丙烯酰胺通过mCPBA环氧化反应制备化合物(I)所需反应时间为10小时左右,而用N-(4-氰基-3-三氟甲基苯基)甲基丙烯酰胺通过次氯酸钠氧化反应制备化合物(Ⅱ)[X=Cl,即化合物(Ⅲ)],所需时间为0.5~1小时,极大地降低了耗能,提高了釜效和生产效率;二是物料成本显著降低,原来普遍用的环氧化试剂间氯过氧苯甲酸(mCPBA)每公斤300元左右,现在使用的次氯酸钠溶液每吨1100元左右,由mCPBA环氧化制备化合物(Ⅰ)的成本为2200元/公斤,采用此专利,由次氯酸钠溶液氧化制备化合物(Ⅱ)的成本为1500元/公斤,并且次氯酸钠溶液广泛用于多个行业,原料很易得。本发明提供一条可适用于大规模生产的制备比卡硫醚中间体的新方法,从而解决比卡鲁胺现有合成技术中存在的安全性差,污染环境,成本高等问题。
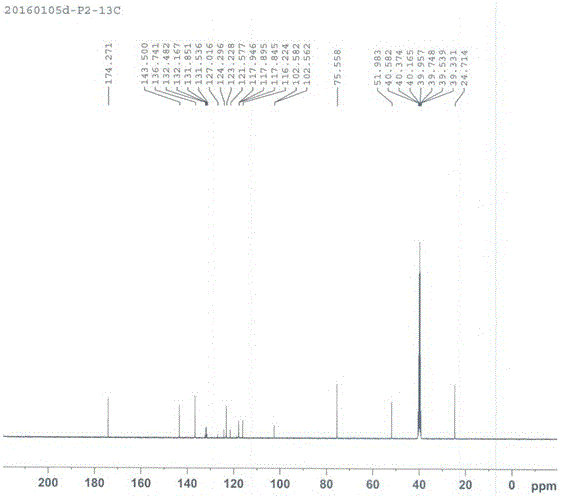
通过1H-NMR(图1),13C-NMR(图2),19F-NMR(图3)和LCMS(图4)对化合物(Ⅲ)进行了结构和纯度表征,详见附图。
附图说明
图1化合物(Ⅲ)的1H-NMR图谱,图中1HNMR(d6-DMSO,400MHz):δ1.46(s,3H),3.71(d,1H, J=11.2),3.94(d,1H, J=11.2),6.42(s,1H),8.12(d,1H,J=8.8),8.34(dd,1H,J=8.8,1.6),8.57(d,1H,J=1.6),10.57(s,1H)。
图2化合物(Ⅲ)的13C-NMR图谱,图中13CNMR(d6-DMSO,100MHz):24.71,51.98,75.56,102.57(d,1C,J=2),116.22,117.91,122.94(q,1C,J=272),123.23,132.01(q,1C,J=32),136.74(q,1C,J=5),143.50,174.27。
图3化合物(Ⅲ)的19F-NMR图谱,图中19FNMR(d6-DMSO,376MHz):-61.21。
图4化合物(Ⅲ)的LCMS图谱,图中ESIMS:m/z305 [M-H]-。
具体实施方式
下面结合实施例对本发明做进一步的详细说明,但不作为对本发明的限制。
实施例1
N-(4-氰基-3-三氟甲基苯基)甲基丙烯酰胺(1g)溶于丙酮15毫升中,冷却至0~5℃,缓慢加入0.58g浓硫酸,搅拌下,滴加10%次氯酸钠溶液5.86g,滴毕搅拌0.5h,反应液中加入乙酸乙酯20毫升和20毫升水,分液,有机相依次用饱和碳酸氢钠溶液和饱和食盐水洗涤,无水硫酸钠干燥,过滤,脱溶得到黄色粗品1.2g,用10毫升甲苯重结晶得到1g类白色晶体,收率83%,HPLC纯度98.02%。
实施例2
N-(4-氰基-3-三氟甲基苯基)甲基丙烯酰胺(1g)溶于甲醇15毫升中,冷却至0~5℃,缓慢加入1.17g浓盐酸,搅拌下,滴加10%次氯酸钠溶液5.86g,滴毕搅拌0.5h,反应液中加入二氯甲烷20毫升和20毫升水,分液,有机相依次用饱和碳酸氢钠溶液和饱和食盐水洗涤,无水硫酸钠干燥,过滤,脱溶得到黄色粗品1.1g,用10毫升甲苯重结晶得到0.95g类白色晶体,收率79%,HPLC纯度97.27%。
实施例3
N-(4-氰基-3-三氟甲基苯基)甲基丙烯酰胺(1g)溶于乙醇25毫升中,冷却至0~5℃,缓慢加入0.58g浓硫酸,搅拌下,滴加10%次氯酸钠溶液5.86g,滴毕搅拌0.5h,反应液中加入乙酸乙酯20毫升和20毫升水,分液,有机相依次用饱和碳酸氢钠溶液和饱和食盐水洗涤,无水硫酸钠干燥,过滤,脱溶得到黄色粗品1.2g,用10毫升甲苯重结晶得到0.91g类白色晶体,收率76%,HPLC纯度97.95%。
实施例4
N-(4-氰基-3-三氟甲基苯基)甲基丙烯酰胺(1g)溶于DMF15毫升中,冷却至0~5℃,缓慢加入1.16g磷酸,搅拌下,滴加10%次氯酸钠溶液5.86g,滴毕搅拌0.5h,反应液中加入乙酸乙酯20毫升和20毫升水,分液,有机相依次用饱和碳酸氢钠溶液和饱和食盐水洗涤,无水硫酸钠干燥,过滤,脱溶得到黄色粗品1.2g,用10毫升甲苯重结晶得到0.95g类白色晶体,收率79%,HPLC纯度97.98%。
实施例5
N-(4-氰基-3-三氟甲基苯基)甲基丙烯酰胺(1g)溶于DMSO15毫升中,冷却至0~5℃,缓慢加入0.58g浓硫酸,搅拌下,滴加10%次氯酸钠溶液10.7g,滴毕搅拌0.5h,反应液中加入乙酸乙酯20毫升和20毫升水,分液,有机相依次用饱和碳酸氢钠溶液和饱和食盐水洗涤,无水硫酸钠干燥,过滤,脱溶得到黄色粗品1.1g,用10毫升甲苯重结晶得到0.90g类白色晶体,收率75%,HPLC纯度97.12%。
实施例6
N-(4-氰基-3-三氟甲基苯基)甲基丙烯酰胺(1g)溶于乙腈15毫升中,冷却至10~15℃,缓慢加入0.58g冰醋酸,搅拌下,滴加10%次氯酸钠溶液5.86g,滴毕搅拌0.5h,反应液中加入乙酸乙酯20毫升和20毫升水,分液,有机相依次用饱和碳酸氢钠溶液和饱和食盐水洗涤,无水硫酸钠干燥,过滤,脱溶得到黄色粗品0.9g,用10毫升甲苯重结晶得到0.7g类白色晶体,收率58%,HPLC纯度96.68 %。
实施例7
N-(4-氰基-3-三氟甲基苯基)甲基丙烯酰胺(100g)溶于丙酮1.5升中,冷却至0~5℃,缓慢加入58g浓硫酸,搅拌下滴加10%次氯酸钠溶液586g,滴毕搅拌1h,反应液中加入乙酸乙酯2升和2升水,分液,有机相依次用饱和碳酸氢钠溶液和饱和食盐水洗涤,无水硫酸钠干燥,过滤,脱溶得到黄色粗品120g,用1升甲苯重结晶得到106g类白色晶体,收率88%,HPLC纯度97.38%,测定其熔点为145.2~147℃。
- 还没有人留言评论。精彩留言会获得点赞!